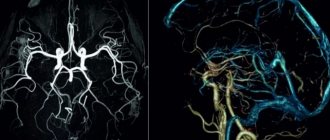
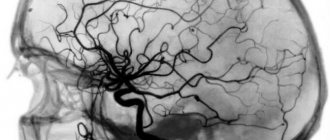
The human brain not only works continuously throughout a person’s life, but also ages along with him. Under the influence of years, a person’s lifestyle and other factors, various destructive processes begin in him. Some of them can seriously affect life in general. For example, one of the unpleasant options is brain atrophy.
It represents changes leading to tissue depletion and loss of organ function. As a rule, when they talk about brain atrophy, they mean that the processes of necrosis of nerve cells have begun, and there is also a rupture of neural connections within groups that are connected either by chemical characteristics or functionally. Against the background of atrophy, the volume of brain tissue begins to decrease. Sometimes this situation is popularly called brain shrinkage.
Related article: Scientists have refuted this. Doesn't higher education affect the youth of the brain? As cells die, serious complications begin to appear. For example, cognitive abilities suffer, that is, a person begins to speak and think worse, he has problems with orientation in space, understanding and logic deteriorate, and abilities such as reasoning, calculation, and learning suffer. Movement problems may also develop. It is worth understanding that the death of neurons does not occur overnight—situations are not uncommon when several decades pass between the onset of the destructive process and the appearance of large damaged areas in the brain.
Destruction in the human brain can be focal, that is, when one fragment is affected, or it can become generalized, when degenerative processes are noted in all areas. Moreover, acute focal atrophy is usually regarded by specialists as a sign of illness or injury. There are also congenital manifestations, which, as a rule, give the first symptoms in childhood.
Why is it developing?
Naturally, many are interested in what are the reasons for the development of such degenerative processes. The risks of brain shrinkage and deterioration increase with older age. Triggers for atrophy are:
- renal failure;
- long-term increased intracranial pressure;
- alcohol abuse and taking illegal drugs;
- infections of the cerebral cortex, such as polio, encephalitis and others;
- traumatic brain injury;
- vascular diseases, such as atherosclerosis, aneurysms;
- metabolic failures;
- the presence of mental illness, in old age this may be Alzheimer's disease, as well as a neurodegenerative condition - Parkinson's disease.
- birth injuries, lack of B vitamins and folic acid;
- heredity.
Let go and don't forget. How the body saves memory when there is poor blood supply Read more
Bipolar Disorder from the Inside Out: What's Wrong with Your Brain and How to Fix It
Share with friends
We have translated for you a chapter from the book “Productive Living with Depression and Bipolar Disorder” by John McManamy. The author himself suffers from bipolar disorder, and at the same time he is confident that a successful and meaningful life with this diagnosis is possible. He devoted years to studying the disease and how to combat it.
In this chapter, the author has compiled current scientific evidence about how nerve cells function, what is wrong with them in a person with bipolar disorder, and how this can be corrected with medication. Here are scientific answers to the questions: why do antidepressants take a few weeks to act, do nerve cells recover, and how exactly do serotonin reuptake inhibitors work?
The material is quite complex - for those who really want to understand how everything works. .
Translation of a chapter from the book “Productive Living with Depression and Bipolar Disorder” by John McManamy, 2006. (John McManamy, Living Well with Depression and Bipolar Disorder)
Neurotransmitters, neurons, and things your psychiatrist doesn't know about
Introduction
In a 1999 report on mental health, the NHS Chief Medical Officer stated: “The brain is the great synthesizer of many biological, psychological and sociocultural phenomena that makes us who we are. It is the result of our genes and our experiences working together.”
What Woody Allen called his "second favorite organ" is nothing less than a 3-pound mass containing approximately 100 billion nerve cells (neurons). They are divided into thousands of different types, each of which in turn forms thousands of connections with other neurons (synapses). There are between 100 trillion and a quadrillion synapses, organized into intricate networks, causing the brain's endless complexity and intricacy. Also, more than half of the 25 thousand genes of the human genome are responsible for its structure.
So what happens when the brain receives a signal? Initially, signals are transmitted from a neuron along a section called an axon, which can end at several terminal sections (terminals). These signals are then picked up by processes (dendrites) coming from other neurons.
Communication between neurons is carried out along countless synapses (gaps) separating axons from dendrites. In these terminal regions of the neuron that send the impulse, special messenger molecules called neurotransmitters are formed. They pass through the synapse and “bind” with receptors on the surface of the neuron receiving the impulse. The neurotransmitter's task is completed when its "message" is delivered through the membrane opening into the neuron.
Neurotransmitters
The brain produces about a hundred neurotransmitters. The three most well-known - serotonin, norepinephrine and dopamine - are classified as monoamines based on their chemical structure. The monoamine hypothesis states that mood disorders are caused by depletion of one or more neurotransmitters. But this theory is reminiscent of the “flat earth” theory: it makes no mention of the latest advances in genetics, neurobiology and brain imaging.
Neurotransmitters are just the first step in mediating the connection between biology and mood. At a 2004 American Psychological Association lecture, Cornell University's Jack Barchas, MD, a pioneer in brain chemistry-behavior research, recounted how, while still an undergraduate, he proposed studying neurotransmitters half a century ago. They answered him: “If you want to study biochemistry, study the liver. It will be years before the processes occurring in the brain are discovered.”
Psychiatry at the time was so far removed from medicine that his mentor challenged these ideas, saying, “How does this prove Freud’s teachings?” Fortunately, Dr. Barkas did not pay attention to these words.
Norepinephrine
Norepinephrine (also called norepinephrine) is produced in neurons from the amino acid tyrosine. Tyrosine is converted to DOPA (dihydroxyphenylalanine) and then to dopamine (see below). Some dopamine is then converted to norepinephrine; it is "packaged" and stored in "packets" called synaptic vesicles. Once norepinephrine is formed by the action of one enzyme, it is in danger of being destroyed by others: for example, monoamine oxidase (MAO), which also breaks down serotonin and dopamine. Monoamine oxidase inhibitors (MAOIs), which inhibit this enzyme and thus prevent the breakdown of monoamines, were the first antidepressants created.
When a neuron is functioning normally, it releases norepinephrine into the synaptic cleft (the space between two neurons). Norepinephrine binds to α1, α2, and β1 receptors on the postsynaptic membrane, the membrane of the neuron on the other side of the synapse. This binding is transmitted to the cell, which activates certain genes that regulate the activity of proteins, which, in turn, determine all the activity of the neuron.
Norepinephrine is most active in a specific part of the brain stem known as the locus coeruleus. It “monitors” external stimuli affecting the body, as well as physiological responses to external stimuli (for example, pain, the “fight or flight” response, etc.). Norepinephrine and the locus coeruleus are also thought to play a role in cognition, mood, emotions, movement, and blood pressure maintenance. Difficulty concentrating, fatigue, apathy and depression are just some of the problems that arise due to disruptions in norepinephrine.
After norepinephrine is released into the synapse and binds to receptors on the postsynaptic membrane, the presynaptic neuron “sucks” some of the remaining neurotransmitter into the cell through a reuptake transporter protein for further packaging into vesicles and subsequent reuse. Tricyclic antidepressants (TCAs) bind to reuptake transport proteins, preventing the neurotransmitter from being “absorbed” and prolonging its circulation and action at the synapse. They also attach to certain serotonin receptors. New antidepressants, reuptake inhibitors, have a similar mechanism of action.
Serotonin
Serotonin, or 5-hydroxytryptamine (5-HT), is synthesized in neurons from the amino acid tryptophan, which is converted to 5-hydroxytryptophan (5-HTP) and then to serotonin. Serotonin is released into the synaptic cleft in the same way as norepinephrine. There are 17 different types of receptors for serotonin, highlighting its importance as a neurotransmitter.
Serotonin is released from the so-called. nuclei of the brain stem, spreading to other formations - the basal ganglia, frontal cortex, hypothalamus, limbic system, spinal cord. Serotonin is also found in the gastrointestinal tract. Not surprising, since this neurotransmitter is responsible for many things: from mood, anxiety levels, sleep, sex drive, to digestion and hunger. Unfortunately, SSRI antidepressants (selective serotonin reuptake inhibitors) increase serotonin concentrations in all synapses. They do not take into account one fact: what has a good effect on mood can negatively affect libido and other body functions. Thus, the word “selective” (that is, acting selectively) in the name of this group of drugs is a complete distortion of the truth.
As with norepinephrine, the presynaptic reuptake transporter protein pulls excess serotonin out of the synapse for subsequent release. Both TCAs and the newer SSRIs and SNRIs (selective serotonin norepinephrine reuptake inhibitors) antidepressants block this protein, leaving more serotonin circulating. However, if this were completely true, then antidepressants would have an immediate effect. In fact, the first two weeks are just “making an impression”; to reveal the full clinical effect, another 2 to 6 weeks of continuous use are required.
One explanation may be that blockade of the reuptake protein decreases the sensitivity of neurons, reducing normal neurotransmitter release for 4 weeks. Another possible reason is that antidepressants also affect the descending cascade of intracellular processes (see below).
Dopamine
Dopamine is to the brain what nuclear power plants are to the electrical grid. We all need energy, but sometimes it can turn into a disaster. It is not surprising that several classes of drugs used in psychiatry and neurology target this neurotransmitter.
For example, antipsychotics (neuroleptics), which work like fire extinguishers, or drugs that “start the engine.” The older generation of MAO inhibitors increased concentrations of dopamine (as well as other monoamines) indirectly, while the atypical antidepressant bupropion achieves this by a different route.
Dopamine is formed from norepinephrine, which, in turn, is formed from tyrosine. There are several dopaminergic nerve pathways in the brain.
The mesolimbic pathway, which runs from the midbrain to the nucleus accumbens, is believed to be involved in pleasure processes and is also responsible for the development of delusions, psychotic states, and drug addiction. Cocaine is notorious for increasing dopamine production, while antipsychotics bind to dopamine D2 receptors, preventing excessive psychoproduction (delusions, hallucinations). Unfortunately, the action of antipsychotics is not limited to just the D2 receptors in the mesolimbic pathway, which leads, as Dr. Steven Stahl said in his book Essential Psychopharmacology of Antipsychotics and Mood Stabilizers, to “too much cost of doing business.”
Dopamine receptor stimulants (agonists) (such as the drug pramipexole for Parkinson's disease) duplicate the effect of natural dopamine. They bind to D2 and D3 receptors in the nigrostriatal dopaminergic pathway from the substantia nigra of the brainstem to the basal ganglia and striatum. The above drug is being clinically tested in patients with depression. A new generation of drugs that stimulate D1 receptors is in development. Dopamine receptor agonists are expected to counteract apathy and cognitive decline.
Glutamate
Glutamate and GABA (γ-aminobutyric acid) are a “yin-yang” pair, as Darryle Schoepp, Ph.D., explained at the 2003 American Psychiatric Association meeting. Both of these neurotransmitters regulate virtually all synaptic functions throughout the brain. , and glutamate is an “excitatory” neurotransmitter, and GABA is an “inhibitory” one. Mood stabilizers (mood stabilizers) are believed to act on either one or the other, or both.
There are 2 types of glutamate receptors - ionotropic (iGluR), which includes NMDA, AMPA and kainate receptors, and metabotropic (mGluR), which controls numerous chemical reactions.
When an NMDA receptor is functioning correctly, glutamate and glycine bind to it, which “opens” an ion channel inside the receptor, allowing calcium ions to enter the neuron. This triggers an intracellular cascade of signaling reactions necessary for cell adaptation and survival.
However, too much glutamate can spell disaster. The mood stabilizer lamotrigine, which has shown effectiveness in treating bipolar depression, is an antiglutamate drug.
GABA
GABA (γ-aminobutyric acid) is formed in the brain from glutamate, glucose and glutamine. It attaches to one of two receptors on the postsynaptic membrane - GABAA or GABAB.
GABAA receptors regulate excitability, anxiety, panic and stress. They are the point of application for benzodiazepine tranquilizers (for example, diazepam), barbiturates, and also alcohol. In people with depression, GABA levels in the cerebrospinal fluid and blood plasma are reduced. Gerard Sanacora, MD, Yale University, used magnetic resonance spectroscopy to measure GABA levels in the brain. He found that people with mild depression have low concentrations of GABA in the occipital cortex, but the depletion of GABA is not as severe as in people with atypical depression. This indicates the possibility of a more detailed approach when diagnosing depression. Dr. Sanacora also compared before and after scans, revealing that GABA levels increased significantly in patients treated with antidepressants or electroconvulsive therapy.
Inside a neuron
Until recently, it was believed that new neurons could not form in the brain. From the point of view of mood disorders, this is a sad fact. Brain imaging and studies of deceased bodies have revealed that in people with depression and bipolar disorder, there is a decrease in the volume of the prefrontal cortex, as well as atrophy of nerve cells and their death. In a classic 1997 study, Dr. Wayne Drevets (National Institute of Mental Health) found that the popliteal region of the prefrontal cortex was 38% smaller than normal in patients with bipolar disorder and 48% smaller in those with depression, regardless of their mental state and treatment.
In experiments conducted by Robert Sapolsky (Stanford University), exposing animals to stress factors led to the death or atrophy of neurons in the hippocampus.
Some neurons were also at risk, more likely to die if exposed to other stressors. One brain component that is deficient during stress is brain-derived neurotrophic factor (BDNF). It is a neuropeptide that plays a key role in the survival and growth of neurons.
Stress increases cortisol levels, which in turn increases the amount of the excitatory neurotransmitter glutamate. This increases the chance of calcium entering the neuron and activates certain calcium-dependent “death enzymes.” Cortisol can also reduce a neuron's ability to store glucose, which supports energy metabolism in the cell. Thus, the cell may not have enough strength to cope with the subsequent crisis.
“Simply put, the cell can’t handle the load,” Dr. Husseini Manji, director of the National Institute of Mental Health’s Anxiety and Mood Disorders Program, explained in a 2003 lecture. Neuronal atrophy and death ensues, reducing their ability to communicate with each other and transmit signals to other cells.
Back in the 1960s. It became known that nerve cells do recover: under favorable conditions, new cells can grow in the brains of animals, and compressed, wrinkled brain cells can return to their normal size and create new intercellular connections - a process called neurogenesis. Most of it occurs in the hippocampus. In 2000, Fred Gage, PhD, Salk Institute, discovered that neurogenesis can also occur in humans.
In 2000, Ron Duman, PhD, and his team at Yale University discovered in rats that antidepressants promote the growth of new cells in the hippocampus. A year later, he published a hypothesis that the same effect might also occur in humans. Dr. Duman and his colleagues were the first to discover that repeated antidepressant treatment stimulates a process known as the cAMP-CREB cascade. CAMP (cyclic adenosine monophosphate) is a signaling molecule found in the DNA sequence of the CREB protein (closer to the 5' end of the DNA). CREB controls the activity of certain genes, including BDNF. It is important that CREB and BDNF play critical roles in neuroplasticity - that is, in the brain’s ability to constantly redistribute memory, “reshape itself” by remembering new information and retaining it.
The cAMP-CREB cascade is also implicated in neurogenesis. Dr. Duman and his team shocked rats' paws to induce behavioral helplessness, leading to long-term suppression of neurogenesis. But when the animals were treated with antidepressants, their behavior returned to normal. Every day, approximately 9,000 new cells are formed in the hippocampus of an adult rodent. Of these, 75–80% become neurons, of which only half survive after 4 weeks. It is estimated that in humans the rate of new cell growth is only 10–20% of that in rodents. According to Dr. Duman, this is still enough to affect hippocampal function.
Meanwhile, a 2000 study led by Dr. Manji found that lithium “significantly increased total brain gray matter volume in people with bipolar disorder.” Using gene microarray—a process that allows researchers to record the interactions of thousands of genes at once—Dr. Manji and his colleagues began experimenting with lithium and valproate on brain cells. To their surprise, they found that two completely different drugs affected the same cellular signaling pathways associated with cell survival or death. In one experiment, levels of a protective protein involved in these signaling pathways (Bcl-2) increased twofold after administration of lithium and valproate. Subsequent experiments on rats revealed that lithium mitigated the effects of an artificially induced stroke and increased the growth of new neurons in the hippocampus. When Dr. Manji asked Dr. Drevets to review his research, it was discovered that patients taking lithium or valproate showed no signs of brain atrophy.
Growing new cells in the brain is just one side of the coin, and most likely not the main one. “More important is the ability to protect and repair damaged brain cells and help them rebuild intercellular connections,” Dr. Manji said. To fully appreciate the benefits of lithium, we must understand that both depression and bipolar disorder are more than just mood disorders. Impaired cognition and behavior may last much longer than a typical mood episode. Although major depressive disorder and bipolar disorder do not belong to the “classical” neurodegenerative (that is, destroying the brain) diseases, such as Alzheimer's and Parkinson's diseases, they are still accompanied by a decrease in the number and shrinkage of brain cells.
“Impressively, Bcl-2 protects against free radicals that can damage brain cells, as well as against the development of Parkinson's disease and possibly the devastation of mood disorders,” Dr. Manji told the National Alliance on Mental Illness annual conference. NAMI) in 2002
Dr. Manji explained that for the past three decades, mental health research has primarily focused on neurotransmitters. But recently, scientists have begun to realize that mental disorders are more complex than previously thought. Nerve cells communicate with each other through neurotransmitters, but they do not travel directly from one cell to another. More precisely, they are simply keys that can open the way to what is happening inside neurons, where all processes take place.
According to Dr. Manji, there are about 10 other potential targets within the nerve cell that we didn't even know were possible 10 years ago. One possible intracellular target is the enzyme protein kinase C (PKC), which is involved in the signaling pathway responsible for neuronal excitability. Dr. Manji's team found that both lithium and valproate have similar effects on the PKC systems and that their effects take days to weeks to begin. Inhibition of protein kinase C, however, can be achieved directly. One small study used tamoxifen, an anticancer drug used for breast cancer. It inhibited (suppressed) protein kinase C. It was found that its use significantly reduced the severity of mania. Trials with a larger sample of patients are planned at the National Institute of Mental Health and Harvard University. “If they are successful,” Dr. Manji said, “we will be able to develop a PKC inhibitor with more advanced properties.”
A review article by Dr. Manji and colleagues from the National Institute of Mental Health (Journal of Biological Psychiatry May 2003) states that the main problem with antidepressant use is the false assumption that our intracellular cascades are intact and will accurately redirect drug-enhanced neurotransmitter activity to the desired points. . In fact, quite the opposite is found: some brain cells mistake the bombardment of neurotransmitters for a kind of violence, which forces them to develop “both trophic and neurochemical support” to restore neuronal connectivity and molecular signal transduction.
Someday, cAMP, Bcl-2, BDNF, and other targets may become as widely known as serotonin is today—not just in academic circles, but also as targets for new drugs to improve our lives.
Other brain cells
There are two fundamentally different groups of brain cells - neurons and glial cells (neuroglia, or simply glia). “Brain glue” is how German scientists unpretentiously called this substance (the word “glia” means “glue” in Greek). As researchers began to study this structure, slowly but surely these “other brain cells” gained their respect.
The story began in the early 1960s, when scientists discovered that the cerebral cortex of rat pups living in an enriched (food, light, etc.) environment contained more glial cells per neuron than those of rat pups growing up in depleted conditions. Apparently, more active cortical neurons required a larger “support network.” As a rule, we remain with the neurons we were born with for the rest of our lives (these cells do not divide). But glia are capable of this. In humans, the number of glial cells and neurons is in a ratio of approximately 9 to 1. This is more than in lower animals.
Two decades after research on the brains of baby rats, four samples of Albert Einstein's brain, each the size of a sugar cube, arrived by mail to one of the researchers, Marian Diamond, PhD, from the University of California, Berkeley. When Dr. Diamond compared regions of the cerebral cortex associated with active cognitive processes with similar regions in 11 control samples, she found that Einstein's brain was literally "filled to the brim" with glial cells.
Advanced neuroimaging techniques confirmed that glia should not have been ignored. Our knowledge was far from perfect, but it now turns out that glia are in a constant, ongoing “dialogue” with the neuron.
Astrocytes are a type of glia that surround the synapses between neurons. Of particular interest is the neurotransmitter glutamate, which binds to receptors on the descending neuron and opens channels on the cell membrane. This allows calcium ions to penetrate and alert the entire “chemical population” inside the cell. Through a series of chemical interactions, astrocytes are able to strengthen signaling using glutamate, releasing it from themselves, or, conversely, weaken it, removing this neurotransmitter from the synapse.
Glutamate (in collaboration with GABA) is a necessary component in mood regulation. When things don't go according to plan, glia always stay put. Some studies on the brains of deceased people who had suffered from major depressive disorder or bipolar disorder while alive showed that they had lower than normal levels of glia in certain regions of the brain. Without glia, the neuron remains practically defenseless; it is unable to resist the “bombardment” of glutamate and the subsequent negative effects of calcium ions.
In a 2004 article in the journal Neuroscientist, Bernhard Mitterauer, MD from the University of Salzburg, proposed the neuronal-glial imbalance hypothesis to explain bipolar affective disorder. The basis of the hypothesis is a different type of astrocyte activity, which involves the release of certain types of proteins into the synapse. These proteins attach to neurotransmitters, preventing them from binding to receptors. According to Dr. Mitterauer, when everything goes according to plan, a certain balance is achieved. But over- or under-secretion of proteins can result in not enough or too many neurotransmitters reaching their target receptors. This leads to negative consequences. This phenomenon (still more theory than fact) may also explain why the sleep-wake cycle is often disrupted in bipolar disorder.
Glial cells also “serve” neurons in a variety of other ways, enhancing the capabilities of our brains. In addition, it is known that glial cells “talk” to each other, but it remains unclear what they “say.” At least after all these years, we are starting to listen to them.
Share with friends
Clinical picture
At the initial stages, symptoms are invisible. However, as it progresses, extremely unpleasant signals may appear:
- convulsions or even epileptic seizures;
- loss of criticality towards one's actions;
- speech impairment;
- problems expressing thoughts;
- memory problems;
- deterioration of the psycho-emotional state - when depression develops, increased irritability is noted, and mood swings are noticeable;
- loss of empathy for other people and much more.
If such symptoms appear that should alert, if not the person himself, then his loved ones, you should contact a specialist to determine the cause of the problem. For example, they conduct a hardware examination - they often talk about MRI and CT diagnostics in order to accurately understand the areas of brain damage, volumes and other important factors. In addition, they may offer examination of cerebral vessels and other options. Next, a decision on therapy is made.
Principles of diagnosing atrophy
The initial stage involves taking an anamnesis, examination, and physical examination. The second stage is clinical and instrumental methods (ultrasound, CT, MRI of the brain, scintigraphy, PET/CT). Damage to the optic nerve is confirmed by ophthalmoscopy, tonometry, contrast CT or MRI angiography.
The best way to detect pathology of the soft tissues of the brain is MRI. The procedure must be performed several times (a month apart) to identify atrophy of varying depth and extent.
Magnetic resonance examination reveals the smallest local lesions and helps to correctly determine the degree of disease progression.
Treatment and prevention
By following simple rules, it is possible to alleviate symptoms and prolong a person’s life. Once the diagnosis is made, it is advisable for the patient to remain in a familiar environment, since stress aggravates the condition. A person needs feasible mental and physical activity.
A balanced diet is important, you need to restore a clear daily routine. It is necessary to give up bad habits. You also need physical activity and mental exercises. A diet for atrophy involves avoiding heavy, unhealthy foods, you should not eat fast food, and drinking alcohol is strictly prohibited. The menu should include nuts, seafood and greens.
Treatment of brain atrophy involves the use of neurostimulants, tranquilizers, antidepressants and sedatives. This disease cannot be completely cured; therapy can only alleviate the symptoms. The choice of remedy depends on the type of atrophy and the type of impaired functions.
With damage to the cerebellar cortex, treatment is required to restore movement. You also need to use drugs that reduce tremor. Sometimes surgery is needed. Sometimes medications are used that improve metabolism and cerebral circulation, ensuring good blood circulation and protection against oxygen starvation.
Source
Symptoms of each stage of the destructive process
Cortical cerebral atrophy goes through five stages of development, each of which has its own symptoms and manifestations:
- The initial stage is characterized by a complete absence of manifestations, but its development occurs rapidly and quickly moves into the next stage.
- At the second stage, communication with others deteriorates very quickly. A person cannot adequately perceive criticism, becomes irritable, conflicts involving him are a normal phenomenon that recurs regularly. Very often the thread of the conversation gets lost.
- The third stage - the patient loses control over his behavior in several stages, which becomes outrageous. Frequent manifestations of despondency or the occurrence of causeless outbursts of anger.
- four : awareness of the essence of events is completely lost. The patient cannot adequately respond to the demands of others.
- Fifth stage - the patient does not take pictures of the events taking place at all, and, accordingly, this does not cause any emotions in him.
Depending on who and what lobe of the brain was affected by atrophy, some speech disorders arise in the early stages, unreasonable euphoria or indifference, lethargy, sexual hyperactivity, and some types of mania appear.