Recently, heart disease is increasingly being diagnosed in childhood. The most common pathology of the heart muscle in children is cardiopathy, a disease of non-infectious origin that manifests itself in changes in heart tissue. How is cardiopathy manifested and treated in children?
Classification of cardiopathy
Cardiopathy is diagnosed in children of different ages. Most often, this disease is detected in the first 2 weeks of a newborn’s life, at 7–9 years old and in adolescence. This pattern is explained by the fact that in the first days of a child’s life they are carefully examined. Particular attention is paid to those children who have poor heredity or prerequisites for congenital pathologies. By the age of 7-9, the child becomes old enough to be able to explain what is bothering him, and the detection of the disease in adolescents is due to a sharp increase in hormonal activity, which leads to the first or secondary exacerbation of hidden pathologies. The types of cardiopathy are described in the table.
| Classification sign | View | Description |
| Pathology detection period | Congenital | Appears due to intrauterine developmental disorders or genetic predisposition |
| Acquired | Occurs as a consequence of the negative impact on the body of various factors (diseases, stress, physical activity) | |
| Causes of the disease | Functional | It manifests itself with systematic physical tension and stress that the child’s body cannot cope with. |
| Secondary | It is a consequence of acute and chronic diseases. | |
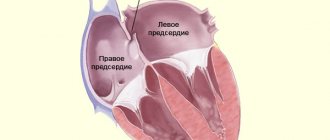
| Changes in the heart muscle | Dilatational | It is expressed in stretching of the muscles of the left ventricle. The walls of the heart do not change in thickness, but due to pathology, the speed of blood flow and the elasticity of the cardiac tissues decrease. |
| Dysplastic | In some places, muscle tissue is replaced by connective tissue, so the heart cannot relax normally. The causes of pathology in children are unknown. | |
| Hypertrophic | The walls of the ventricles of the heart thicken, the volume of the heart chamber decreases. This makes it difficult for blood to flow in and out of the heart. |
Symptoms of cardiopathy
Diseases of the cardiovascular system can be asymptomatic for a long time. Then signs specific to cardiopathy appear. The characteristics of the disease consist of gradually increasing symptoms. Patients present the following complaints:
- dyspnea;
- pain in the heart area;
- swelling of the lower extremities;
- cyanosis;
- increased sweating;
- tachycardia;
- cough;
- an increase in the size of the spleen and liver;
- fast fatiguability;
The feeling of lack of air is associated with stagnation of blood in the lungs. Shortness of breath first occurs with excessive physical exertion. Then patients notice that they begin to choke even with slight overexertion. Symptoms gradually increase and are observed at rest.
Some patients report pain in the heart area as one of the first symptoms. Cardiopathy of the dysplastic form is characterized by stabbing or squeezing.
Against the background of heart failure, blood flow in the vascular bed slows down, and this is manifested by swelling of the lower extremities. They first appear in the evening. As the disease progresses, swelling becomes a constant symptom. Violation of the outflow of fluid leads to its accumulation in the abdominal cavity (ascites).
Due to stagnation of blood in the veins, a bluish tint appears. Most often this is observed on the skin of the fingertips, earlobes, lips, and nasolabial triangle. Enlargement of the liver and spleen is observed when blood flow through the vessels is disrupted.
Symptoms of pathology in children
In infancy, it is difficult to notice cardiac abnormalities. Kids cannot explain what causes them discomfort. You can suspect cardiopathy in an infant based on the following symptoms:
- with strong crying, the area of the nasolabial triangle turns blue;
- the rhythm of breathing is lost;
- the baby gets tired quickly.
Older children experience more extensive symptoms. With cardiopathy, children often complain of pain in the area of the heart or chest. The disease is also accompanied by:
- high fatigue;
- shortness of breath;
- dizziness and fainting;
- pale skin;
- increased sweating.
The manifestation of symptoms is due to the fact that due to improper functioning of the heart, the delivery of blood to other organs is disrupted. With some types of disease, health worsens even with such physical activity that a healthy child does not feel.
The dilated form of cardiopathy, in the absence of timely treatment, leads to the appearance of a bulge in the area of the heart. This occurs because the heart muscle expands significantly and causes displacement of nearby tissue.
The most striking sign of functional cardiopathy is cardiac arrhythmia. If physical activity is not reduced, it leads to a significant deterioration in heart condition.
Secondary cardiopathy is accompanied by the same symptoms that are inherent in the underlying disease. In this regard, secondary cardiopathy is difficult to recognize.
Based on our own experience and literature data, a description of the genetic nature of hypertrophic cardiomyopathy, its clinical picture, and the results of instrumental studies is given. Treatment is being discussed. Criteria for poor prognosis are given.
Hypertrophic cardiomyopathy in children
Based on our experience and the literature describes the genetic nature of hypertrophic cardiomyopathy, its clinical presentation, results of instrumental studies. It is discuss the treatment. Some criteria of poor prognosis were held.
Hypertrophic cardiomyopathy (HCM) is a structural and functional lesion of the myocardium with an increase in its thickness (mass) with a known or suspected genetic defect of the cardiomyocyte sarcomere protein, inherited autosomal dominantly with varying penetrance and expressivity in the absence of systemic hypertension, storage diseases, hypothyroidism, atherosclerotic coronary sclerosis, valve defects, congenital heart defects and other conditions that could explain the increase in myocardial mass. Modern possibilities for the treatment of HCM with long-term support of a sufficiently high quality of life have actualized the problems of early diagnosis and adequate choice of patient management tactics [1, 2].
Etiology.HCM is inherited according to Mendelian laws in an autosomal dominant manner. The form of the disease in probands and their closest relatives is the same in 60% of cases. The genes responsible for the disease are localized on chromosome 6 (15 genes) and on the long arm of chromosome 14 [3]. Gene changes are represented by more than 400 missense mutations responsible for the regulatory, structural or functional characteristics of thin or thick filaments of cardiac sarcomeres (β-myosin heavy chain, actin, titin, myosin-binding protein C, tropomyosin T, etc.). The number of mutations itself determines the clinical variability of HCM. More than half of all genetically identified cases of HCM are determined by mutations in 3 genes that determine defects in the β-myosin heavy chain, myosin-binding protein C, and cardiac troponin T. Another 13 genes are responsible for infrequent cases of defects in titin, α-tropomyosin, α-actin, and cordial troponin I, myosin light chain. But among people with clinically and instrumentally diagnosed HCM and subjected to molecular genetic testing, gene abnormalities were detected only in 50-80% of cases [4, 5]. This means that in 20-50% we are dealing with mutations that have not yet been identified. It is likely that different populations will have their own mutations. Thus, it is suggested that the Russian population may be characterized by a missense mutation in the β-MHC gene.
The polymorphism of the clinical picture is explained by the insufficiently studied interaction of various genes (mutation MYH7 - pronounced hypertrophy, early onset and severe course; mutation TNNT2 - moderate hypertrophy; mutation MYBPC3 - incomplete penetrance and relatively late onset), gender, and external factors.
Pathophysiology . The gene defect leads to discoordination of the activity of myofibrils and subsequent fibrosis and hypertrophy of the myocardium. Chaotic cellular structure already occurs in areas without signs of hypertrophy and which are an arrhythmogenic substrate of ventricular tachycardia and fibrillation. At the same time, the intima of the intramural coronary arteries thickens, oxygen transport into the thickened myocardium is disrupted, ischemia, cell death, and sclerosis occur. The walls of the ventricles thicken, but the internal size remains unchanged or even decreases. Systolic function does not change for a long time, myocardial rigidity increases sharply, end-diastolic pressure increases, and blood return is inhibited. Hypertrophied myocardium can narrow the LV outflow tract. The degree of obstruction varies depending on the thickness of the interventricular septum, the location of the mitral annulus, anterior systolic movement of the mitral valve leaflets, and the intensity of myocardial contraction (therefore, digitalis preparations are contraindicated). The degree of obstruction does not correlate with the risk of sudden death. Progression of the disease leads to ventricular dilatation.
An experiment on animals with a positive HCM genotype but a negative phenotype showed that the administration of calcium channel blockers before the development of hypertrophy can prevent the disease [6]. Such experimental therapy is also carried out in people who are presymptomatic carriers of the HCM genotype.
The prevalence of HCM in children is 3-5: 1 million, median age is 7 years, 1/3 of all cases are diagnosed before the age of 1 year. In 25-58% of 1st degree relatives, echographic signs of HCM are detected. The incidence of HCM is approximately the same in different populations, although atypical variants of HCM are more common in Asia. Among children, there are gender differences in the incidence of HCM, but in adulthood, the disease is more often detected in men. The differences are explained by hormonal factors, environmental influences, more severe symptoms or a greater degree of LV outflow tract obstruction in men. All this determines more frequent studies leading to the diagnosis of HCM.
Clinical picture. There may be no complaints, and in these cases the diagnosis of HCM is the result of random echocardiographic studies. According to the results of our own studies (84 patients), indications of fainting, shortness of breath, a feeling of interruptions in the heart, etc. are more common. According to the results of 24-hour blood pressure monitoring, a tendency to bradycardia is recorded, apparently due to a decrease in LV ejection.
Shortness of breath is one of the earliest symptoms and occurs in 90% of patients. Dyspnea is caused by high rigidity of the thickened myocardium, increased end-diastolic pressure and transfer of pressure to the pulmonary circulation.
Syncope and presyncope are common in HCM, especially in children and adolescents with small LV diameter and ventricular tachycardia or sick sinus syndrome or sinus node dysfunction on 24-hour monitoring. In our group of patients they occurred in 27%, more often in the obstructive form of HCM. The presence of fainting is an indicator of a high probability of sudden death. Presyncope, which occurs during a sudden transition to a vertical position, can be caused by transient atrial or ventricular tachyarrhythmia and is considered a predictor of sudden death.
Dizziness is common in children with HCM with a high gradient in the left ventricular outflow tract and is aggravated by physical activity or hypovolemia (profuse sweating in the heat), rapid transition to an upright position, or a Valsalva reaction during bowel movements. Dizziness can occur with rhythm disturbances with decreased cerebral perfusion. Short-term episodes of arrhythmia are manifested by presyncope and dizziness, while persistent arrhythmias lead to fainting and sudden death.
Anginal pain is typical for children with HCM. There is no atherosclerotic coronary sclerosis. The symptoms of myocardial ischemia are explained by the increased need of the thickened myocardium for oxygen, which, if diastole is disturbed, leads to subendocardial hypoxia, especially during physical exertion. The feeling of interruptions in the heart is explained by premature atrial or ventricular contractions, intermittent atrioventricular block, supraventricular or ventricular tachycardia. Undiagnosed ventricular tachycardia is an indicator of a high risk of sudden death.
Orthopnea and paroxysmal nocturnal dyspnea are signs of heart failure in severe types of HCM with venous congestion in the lungs. Congestive heart failure in children occurs in only 10% of cases (much less often than in adults) and mainly before the age of 1 year. It is characteristic of patients with the most pronounced thickening of the myocardium. Congestive heart failure is the result of two damaging factors: high myocardial stiffness and subendocardial ischemia.
The apical impulse is diffuse, elevating, shifted to the left. A double impulse is typical, the upper part of which is formed by increased contraction of the left atrium. The most typical for HCM is the triple apical impulse, the last part of which is caused by isometric contraction of the ventricle. But this option is extremely rare.
Venous jugular pulse - a pronounced wave “a” on the venogram: a sharp increase in end-diastolic pressure in the left ventricle with an increase in pressure in the pulmonary circulation and thickening of the interventricular septum with a decrease in the cavity of the right ventricle, ultimately with inhibition of inflow to it.
The carotid pulse is high, fast and short, which is explained by the high speed of blood flow through the narrowed LV outflow tract. In some cases, the carotid pulse appears as a successive double pulsation of the carotid artery: 1st peak - as described earlier, 2nd - a low rise of the artery in late systole after the fall of the gradient.
Auscultation of the first tone is not changed, the second is usually split, and with a very high gradient it is paradoxical. A gallop rhythm or a third sound is often heard, but in HCM it does not have the same prognostic value as in valvular aortic stenosis. The IV tone is often determined due to forced atrial systole, overcoming resistance to blood flow into the LV. The diamond-shaped systolic ejection murmur (crescendo-decrescendo) is best heard at the point between the apex and the left edge of the sternum, radiates suprasternally, but is not transmitted to the carotid arteries. The intensity of the noise is determined by the subaortic gradient in the LV outflow tract. Since the degree of pressure gradient varies depending on the obstruction and the volume of inflowing blood, the intensity of the noise is not constant. The noise decreases with increasing preload: Valsalva maneuver, Müller maneuver (after forced exhalation, the inhalation movement is performed with the mouth and nose closed (subatmospheric pressure occurs in the chest and lungs = inverted Valsalva maneuver), squatting position. The gradient increases and the noise becomes louder as it decreases preload (nitrates, diuretics, standing) or decrease after exercise (vasodilators). A holosystolic murmur occurs with anterior systolic movement of the mitral valve and a very high gradient in the LV outflow tract. In 10% of children with HCM, a diastolic murmur is heard decrescendo regurgitation at the aortic valve, but with Doppler ultrasound, minimal and moderate aortic regurgitation is detected in 33%.
Additional research. Electrocardiography most often reveals signs of LV hypertrophy, deviation of the electrical axis of the heart, conduction disturbances, sinus bradycardia (33%) with ectopic atrial rhythm and atrial enlargement. With adenosine monophosphate mutation activated PRKAG2, HCM is combined with congenital Wolf-Parkinson-White syndrome and conduction disorders.
Deep Q waves in the precordial leads (40%), atrial fibrillation (a poor prognostic sign) and P wave abnormalities are relatively common. The size of the Q wave directly correlates with the thickness of the myocardium of the posterior wall of the LV (Fig. 1).
Figure 1. Electrocardiogram of patient N., 5 years old, with obstructive form of HCM. There is a pathologically deep Q wave in leads I, AVL, V5-V6. The voltage of the R wave in leads II, III, AVF, V6 is reduced. The thickness of the interventricular septum according to echocardiography is 1.5 cm, the posterior wall of the left ventricle is 0.7 cm.
The Q wave in leads II, III, aVF was recorded mainly in patients with moderate isolated myocardial hypertrophy. In severe progressive HCM, in addition to leads II, III, aVF, the Q wave was recorded in leads V4-V6; in cases of combined hypertrophy of the LV and right ventricular myocardium - in leads I, II, aVL, V4-V6. It can be assumed that the pathological Q wave is not only an isolated sign of hypertrophy of the interventricular septum, but also an indirect sign of the characteristics of LV hypertrophy. Changes in the repolarization process are typical: a decrease in the amplitude of the T wave (21% of those examined), its pronounced inversion (61%) up to giant negative T waves in leads V1-V6. Patients with the obstructive form of HCM are characterized by atrioventricular blockades.
With Holter monitoring, rhythm and conduction disturbances are recorded much more often than with simultaneous ECG recording.
There are major and minor ECG criteria for HCM. Major criteria include: 1) Q wave change (>40 ms or 1/3R) in at least 2 leads and 2) T wave inversion (≥3 mm) in at least 2 leads. Minor: 1) left atrium enlargement (Pv1), 2) PR<120 ms, 3) His bundle block or hemiblock (or interventricular conduction disorder: QRS≥120 ms), 4) deep wave Sv2, 5) reoplarization disorders. ECG criteria are nonspecific and must be combined with ultrasound data. For adults, echographic criteria are stable due to the completeness of the body’s development. For a child, the concept of normal is more blurred. Therefore, in children it is advisable to use the ratio of the thickness of the interventricular septum and the posterior wall of the LV. The latter is normally always thicker than the interventricular septum.
2D echocardiography is a leading diagnostic technique. The interventricular septum is thickened relative to the posterior wall of the LV. The internal diameter of the ventricle is at the lower limit of normal or reduced (Fig. 2).
Figure 2. Echogram along the long axis of the LV with asymmetric hypertrophy of the interventricular septum. The interventricular septum is sharply thickened and overlaps the LV outflow tract by almost half. 1 and 2 – aortic wall, 3 – interventricular septum, 4 – posterior wall of the LV.
The smaller the LV diameter, the higher the likelihood of fainting. With mitral regurgitation and/or low LV compliance, the left atrium cavity expands. A typical sign of HCM is anterior systolic motion of the mitral valve. It is explained by the displacement of the mitral ring into the outflow tract of the LV due to thickening of the interventricular septum and the shift of the valve leaflets to the septum (the pressure behind the closed leaflets of the mitral valve is high, and in front of them in the outflow tract, according to Bernoulli’s law, the pressure in the fast fluid flow is low) during systole (echographic Venturi phenomenon). Doppler ultrasonography is used to determine the rate of blood ejection in the LV outflow tract or the rate of blood flow in diastole through the left atrioventricular orifice. In the latter case, the velocity in the rapid filling phase (VEF) is reduced, the peak velocity in the systole phase of the left atrium (VA) relative to VEF is increased. This pathological relationship is also found in genotypically positive but asymptomatic patients.
The X-ray picture varies from unchanged to enlarged cardiac shadow. The left atrial arch bulges with high LV stiffness and mitral regurgitation.
Magnetic resonance imaging makes it possible to clarify the degree of obstruction and the anatomy of cardiac structures and reveals myocardial fibrosis, the prevalence of which directly correlates with the risk of sudden death.
Cardiac catheterization is performed to determine the degree of obstruction, the state of diastolic function, the interventricular septum and the coronary arteries.
Laboratory tests are nonspecific and are required only when secondary forms of myocardial thickening are excluded. Genetic testing most often (80% of HCM cases) reveals mutations in 9 sarcomeric genes MYH7, MYBPC3, TNNT2, TNNI3, TNNC1, TPM1, ACTC, MYL2, andMYL3 and in the CAV3 regulator gene.
Histologically, the myocardium is hypertrophied, myocardiocytes and myofibrils are arranged randomly, and fields of fibrosis are visible. In 80% of patients, the walls of the intramural branches of the coronary arteries are thickened, and the lumen is unevenly narrowed. These changes are most often recorded in the interventricular septum and are accompanied by fields of fibrosis. Along with the listed changes, even in supposedly unchanged areas of the myocardium, defects in calcium transport, disturbances in glycogen metabolism and other transformations are found that adversely affect the prognosis of the disease. This point of view can be confirmed by the fact that in 60% of those who died from HCM, according to light microscopy, 10-15% of the myocardial mass was changed, while for a fatal outcome in myocardial infarction or post-infarction cardiosclerosis, damage to at least 50% of the myocardial mass is required [7 , 8].
Doctor's tactics. After identifying myocardial thickening, it is necessary to carry out differential diagnosis with secondary causes of cardiac muscle hypertrophy:
- "the heart of an athlete" Long-term exercise can lead to myocardial hypertrophy, but with an appropriate history, concentric myocardial hypertrophy with expansion of the LV cavity is detected, which is not typical for HCM;
- aortic stenosis, aortic, subvalvular, coarctation of the aorta;
- restrictive cardiomyopathy;
- storage diseases (glycogenosis, mucopolysaccharidosis, amyloidosis, etc.);
- arterial hypertension (including neonatal);
- congenital hypothyroidism, newborn from a mother with diabetes mellitus;
- neuromuscular diseases;
- ventricular fibrillation.
General recommendations, monitoring physical activity and dynamics of disease symptoms. There is no special diet. Parents and patients should be warned about limiting physical activity, abolishing anaerobic exercise, and avoiding sports (especially in cases of arrhythmias and familial cases of sudden death). Avoid dehydration, control body weight. In dynamics, the thickness of the myocardium (myocardial mass), indicators of LV diastolic function, the diameter of its outflow tract, the pressure gradient, the presence of anterior systolic movement of the mitral valve, and arrhythmias are assessed. In the absence of emergency indications, an ECG is performed every 6 months, an ultrasound examination of the heart - annually, during puberty - every 6 months. If symptoms of HCM or signs of obstruction occur, it is advisable to initiate therapy with calcium channel blockers or β-blockers. In case of severe obstruction, together with surgeons, determine the indications for myectomy (or alcohol ablation) and pacemaker implantation. Prevention of bacterial endocarditis with antibiotics is not indicated. Diuretics, inotropic drugs, nitrates and sympathomimetics are contraindicated. Rule out digitalis unless there is uncontrolled atrial fibrillation.
Controlling risk factors for sudden death is the goal of any therapy for HCM. There are no uniform recommendations. Prognostic factors include familial cases of sudden death, familial variants of HCM, anamnestic indications of periods of asystole, unexplained fainting, pressure gradient (disputed), arrhythmias, thickness of the interventricular septum.
Drug therapy. The drugs of choice are β-blockers and calcium channel blockers. For heart rhythm disturbances, amiodarone and other antiarrhythmic drugs are prescribed. β-blockers reduce the pressure gradient in the LV outflow tract and optimize LV compliance. The effect of treatment was recorded in ½ patients. There is no evidence that β-blockers reduce the risk of sudden death. An alternative to β-blockers are calcium channel blockers. They improve diastolic function and reduce the pressure gradient by reducing LV contractility.
Propranolol (Inderal) is a non-selective β-blocker, the dose is adjusted according to the clinical effect. Atenolol (tenormin) is a selective β1 receptor blocker with minimal effect on β2 receptors. Efficiency is higher than that of propranolol.
Verapamil (isoptin) blocks slow calcium channels in vascular smooth muscle cells and myocardiocytes. The dose is selected individually, maximum effectiveness has been recorded during physical activity.
Amiodarone (cordarone), an effective antiarrhythmic drug, has several points of application. If long QT syndrome is excluded, it is the drug of choice in the treatment of life-threatening ventricular arrhythmias refractory to β-blockers.
Implantation of a pacemaker [7] leads to an improvement in the course of HCM and a reduction in pharmacological load.
LV myectomy with a gradient of 50 mm. rt. Art. regardless of whether it was recorded at rest or during provocative tests. In most cases, the positive dynamics of symptoms persists for 5 years. There is no complete correlation between the success of myectomy and the likelihood of sudden death. Catheter transvenous alcohol ablation of the interventricular septum is more dangerous than open surgery. It is fraught with irreversible atrioventricular block, myocardial infarction, necrosis of the interventricular septum with its iatrogenic defect [9].
Forecast. Sudden death may be the first and fatal manifestation of HCM in previously asymptomatic patients. In children and adolescents, sudden death is provoked by physical activity and sports activities. The maximum mortality rate was recorded in children with HCM diagnosed before the age of 1 year. In the group of children over 1 year of age, mortality is 1-1.4% per year [10, 11]. In 80-85% of cases, sudden death is the result of atrial flutter. Ventricular flutter develops after atrial flutter or fibrillation, ventricular tachycardia, supraventricular tachycardia in combination with Wolff-Parkinson-White syndrome, or catastrophically low cardiac output with hemodynamic collapse. The most important condition for preventing sudden death is early diagnosis of HCM, which allows changing lifestyle, physical activity, prescribing medication, determining indications for surgical treatment or implantation of a cardioverter defibrillator. Taking into account the autosomal dominant type of inheritance, in order to prevent familial cases of sudden death, examination of relatives is required.
Other complications of HCM include infective endocarditis, heart failure, arrhythmias, and atrial fibrillation with intramural thrombosis.
The criteria for poor prognosis and progressive course of HCM in children are:
- family history of sudden death;
- formation of a QS type complex and increasing conduction disturbance along the left bundle branch;
- high grade ventricular arrhythmia;
- atrial fibrillation;
- sick sinus syndrome;
- distal atrioventricular block 1-2 degrees;
- initially low rates of heart rate variability or their decrease by 30% during observation;
- decreased heart rate variability while taking β-blockers.
Conclusion
Hypertrophic cardiomyopathy is a genetically determined disease that rarely manifests itself in childhood and is more often detected in adults. Myocardial hypertrophy is often asymmetrical. Although any sector of the LV can be affected, the interventricular septum often becomes thickened with obstruction of the LV outflow tract. Systolic functions remain intact for a long time, but compliance suffers early. HCM is a chronic disease with a variable clinical picture from the complete absence of clinical symptoms to significant limitation of vital activity with sudden death. HCM is the leading cause of sudden death during exercise in adolescents. The pediatrician is required to organize interdisciplinary and interprofessional support for patients with the choice of therapeutic or surgical tactics, determine the degree of risk of sudden death and its prevention, and screening examinations of relatives.
V.M. Delyagin, E.A. Tikhomirova, A. Urazbagambetov
Federal Scientific and Clinical Center for Pediatric Hematology, Oncology and Immunology, Moscow
Delyagin Vasily Mikhailovich - Doctor of Medical Sciences, Chief Researcher of the Department of Rare Diseases, Head of the Department of Functional Diagnostics
Literature:
1. Maron B. Hypertrophic cardiomyopathy. A systematic review // J. Am. M.As. - 2002. - Vol. 287. - P. 1308-1320.
2. Maron B., McKenna W., Danielsen W. et. al. American College of Cardiology // Eur. Heart J. - 2003. - Vol. 24. - P. 1665-1691.
3. Jarcho J., McKenna W., Pare J. et. al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1 // New Engl. J. Med. - 1989. - Vol. 321. – P. 1372-1378.
4. Bos J., Towbin J., Ackerman M. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy // J. Am. Coll. Cardiology. - 2009. - Vol. 54. - P. 201-211.
5. Kim L., Devereux R., Basson C. Impact of genetic insight into Mendelian disease on cardiovascular clinical practice // Circulation. - 2011. - Vol.123. — P. 544-550.
6. Semsarian C., Ahmad I., Giewat M. et. al. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model // J. Clin. Invest. - 2002. - Vol. 109. - P. 1013-1020.
7. Epstein A., DiMarco J., Ellenbogen K., Estes N. et. al. ACC/AHA/HRS 2008 Guidelines for Device-Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology // J. Am. Col. Cardiol. - 2008. - Vol. 51. - P. 62.
8. Siegenthaler W. Differential diagnosis innerer Krankheiten // Neubearbeitete Auflage. - Stuttgart: Georg Thieme Verlag, 1993.
9. Sorajja P., Valeti U., Nishimura R., Ommen S. et. al. Outcome of alcohol septal ablation for obstructive hypertrophic cardiomyopathy // Circulation. - 2008. - Vol. 118. - P. 131-139.
10. Tikhomirova E.A. Criteria for predicting the course and outcome of hypertrophic cardiomyopathy in children: abstract of thesis. ... Ph.D. - M. - 2007. - 27 p.
11. Colan S., Lipshultz S., Lowe A. et. al. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry // Circulation. - 2007. - Vol. 115. - P. 773-781.
Diagnostic methods
If cardiopathy is suspected in a child, the doctor conducts a careful examination and asks the parents about the presence of symptoms. The initial examination is performed by a pediatrician, but a more thorough diagnosis is carried out under the supervision of a cardiologist.
Pediatric cardiology uses the following methods to diagnose cardiac pathologies:
- Echocardiography. The study consists of examining the chest using an ultrasound machine. With its help, the specialist determines the parameters of the heart muscle and its structural elements. Another name for the procedure is cardiac ultrasound. The procedure allows you to identify dysplastic pathology without the use of other methods.
- ECG. An electrocardiogram records the electrical signals produced by the heart. The device records the heart rate, describes the physical condition of the heart, and indicates the presence of arrhythmic movements.
- Phonocardiography. This method is in addition to ultrasound and ECG. It allows you to record the noise that appears during the work of the heart muscle.
Additionally, the child may be prescribed blood tests to determine genetic abnormalities. In some cases, it is necessary to determine the cause of the pathology. To do this, he is given a referral for a general blood test, CT scan, and MRI.
Characteristics of the disease
The main function of the heart muscle is to contract rhythmically, which pushes blood through the vessels and fills the cavities of the organ. This ability is provided by cardiomyocytes. Their continuous functioning maintains metabolism at the level necessary for work.
Under the influence of unfavorable factors, over time the processes presented are disrupted. This is manifested by the formation of structural changes, which ends with a weakening of contractility.
Our capabilities
If atypical ventricular chord is suspected or previously identified, the following examinations are recommended:
- Echocardiography is the most informative method for diagnosing various heart pathologies. During the study, the doctor evaluates the structure and contractility of the myocardium, the functioning of the valves, the movement of blood in the chambers of the heart and other indicators.
- A resting ECG is a basic test for assessing heart rhythm. Does not require special training.
You can undergo all diagnostic tests at any KinderKlinik department with subsequent consultation with a specialist.
Need some advice?
Sign up
Callback form
Application has been sent, we will contact you soon
Treatment of dysplastic cardiopathy
It is most appropriate to begin treatment with a drug-free approach. If your doctor allows dosed physical activity, you can play sports subject to strict adherence to the recommendations. Particular importance is given to walks in the fresh air and proper nutrition.
You should eliminate junk food from your diet, drink clean water and limit salt. You need to eat fresh fruits and vegetables every day. This not only has a beneficial effect on the cardiovascular system, but also on the immune system and digestive tract.
The patient is indicated for physiotherapy. One of them for dysplastic cardiopathy is interference therapy. You can start doing it only from the age of 10-13 years, depending on contraindications. Treatment is performed using the AIT device. It emits currents after two electrodes are applied to the skin. They are transformed in the thickness of tissues from mid-frequency to low-frequency.
The described transformation occurs when the first ones are superimposed on each other. The process is called interference, which is where the procedure gets its name. The generated impulses improve blood supply to organs and tissues. The functioning of the patient’s nervous system is normalized. Anxiety disappears, the frequency of awakenings during the night decreases.
Such frequencies allow you to reduce muscle tone and dilate blood vessels. Under their influence, after several sessions, pain becomes less pronounced and swelling in the legs is less noticeable. We must not forget about contraindications to interference therapy:
- oncological diseases;
- high body temperature;
- inflammatory diseases in the acute stage;
- tuberculosis;
- violation of blood clotting processes.
Depending on the severity of the condition, the patient is offered drug therapy. It is recommended to start with the introduction of herbal sedatives (valerian tincture) to reduce anxiety and fear. It is most advisable to introduce such drugs into the treatment regimen for children.
Among the tablets indicated for children with dysplastic cardiopathy are Anaprilin and Verapamil. Some patients are prescribed hormone therapy. The positive dynamics of treatment can be traced from the clinical picture.
Older children and adults report a decrease in the severity of symptoms of the disease and improved sleep. For control, electrocardiography (ECG) film must be removed. The required dose is individual, depending on the manifestations, age and the presence of concomitant pathology.
The appearance and development of the disease
Dysplastic cardiopathy is a disease associated with impaired myocardial development. Patients experience replacement of normal heart tissue with fibers with a low degree of elasticity. They lose the ability to fully perform their functions. Against the background of such changes, a characteristic clinical picture arises.
The diagnosis is made if there is a history of mitral valve prolapse (MVP) or an extra chord in the left ventricle. The ICD code for dysplasia is available only in the CIS countries. The diseases indicated above are indicated by separate codes. It is customary to add to it the pathology that led to this outcome. Before prolapse, they most often write a minor anomaly of the heart, which was the cause of the defect.
In most cases, the diagnosis is made in the maternity hospital. Dysplastic cardiopathy in children is a group of diseases of non-inflammatory origin. The fibers of the heart muscle, which should be elastic, gradually change. In their place, rough connective tissue appears, devoid of such ability.
It is the elasticity of the fibers that is considered one of the most important features for ensuring complete contraction of the myocardium.
Cardiopathy leads to the appearance of organic and functional disorders. The following changes in the heart are distinguished:
- valve stenosis;
- anastomosis between arteries;
- disruption of the conduction of impulses through the myocardium;
- displacement of the heart axis;
- disruption of developmental processes in one of the ventricles.
After rheumatism, dysplastic cardiopathy develops in children. The characteristic of the disease is the appearance of symptoms of varying severity. They are associated with a violation of integrity, which occurs against the background of high load in the myocardium.
When connective tissue appears in place of normal elastic fibers, the work of the myocardium changes. The walls of the ventricles retain the ability to contract, but cannot relax at the same level as before. The changed structure makes cardiomyocytes stiff and less elastic.
The described characteristics of the pathological process indicate a gradual impairment of cardiac function. When there is an imbalance, the atria become enlarged, but the ventricles remain normal. The dysplastic process leads to limited blood flow. A child most often falls ill under the influence of various factors while still in the womb, and an adult – after certain illnesses.
Complications
A minor anomaly in the development of the heart (cardiopathy) is dangerous not only for its manifestations, but also for its complications. They can form due to late diagnosis and lack of treatment. With dysplastic cardiopathy, the following consequences are possible:
- angina pectoris;
- cardiac ischemia;
- arterial hypertension;
- pericarditis;
- pulmonary edema;
- arrhythmia;
- thrombosis;
- chronic heart failure.
The most common complication is arrhythmia. It appears in almost 10% of the total number of sick children. Cardiopathy leads to disruption of the normal conduction of electrical impulses in cardiomyocytes. As a result, heartbeats become irregular. Most often there is an acceleration in the number of beats per minute.
When the ventricles dilate, when the disease lasts for a long time, the blood in the cavities stagnates. Conditions are created for the formation of clots.
The greatest danger is represented by blood clots that enter the bloodstream (emboli) and over time they can penetrate into narrow-diameter vessels. This feature is characteristic of lung and brain tissue. Blockage of the lumen in them will lead to throboembolism.