Small ultrasound markers of congenital pathology
Ultrasound scanning during pregnancy is carried out three times in each trimester. The most important is the first prenatal screening, on the basis of which chromosomal abnormalities in the fetus can be detected in a timely manner. Gross anatomical defects are the easiest to identify: it is much more difficult when the doctor sees minor defects. Small ultrasound markers of chromosomal abnormalities include:
- Hyperechoic focus in the chambers of the heart (GEF in the ventricles of the fetal heart);
- Vascular cysts;
- Dilated hyperechoic intestine;
- Enlargement of the renal pelvis;
- Shortening of the limb bones (femur or humerus);
- Thickening of the neck fold;
- Ventriculomegaly (enlargement of brain cavities).
None of these signs can indicate the presence of congenital anomalies, but the ultrasound diagnostic doctor cannot guarantee the absence of developmental defects. GEF in the ventricles of the fetal heart is a risk factor for the possible development of pathology and a reason for careful assessment of the intrauterine growth and development of the baby.
Publications in the media
Guillain–Barré syndrome is a post-infectious demyelinating polyneuropathy, the most common demyelinating lesion. It is characterized by peripheral paralysis of the muscles of the limbs and protein-cell dissociation in the cerebrospinal fluid while maintaining superficial sensitivity. May have an ascending nature involving the muscles of the face, pharynx, and larynx (ascending Landry's palsy). Frequency: 0.6–1.9 cases per 100,000 population. The predominant gender is male.
Etiology. There is an opinion about the autoimmune nature of the disease. Develops after or during the following conditions: • Infectious diseases •• Upper respiratory tract infections •• Infectious mononucleosis •• CMV infection •• Herpes infection •• Influenza A •• Mycoplasma infection •• Mumps •• HIV infection • Lymphoma (especially Hodgken) • Vaccination, serum sickness • Surgery.
Pathogenesis. Selective demyelination of the spinal cord roots, apparently of an autoimmune nature. Against the background of immune disorders, edema, inflammatory cell infiltration and diffuse primary segmental demyelination occur primarily in the anterior roots and proximal parts of the spinal nerves, plexuses, limb nerves and autonomic ganglia.
Pathomorphology • Segmental demyelination of peripheral nerves, in severe cases - axonal degeneration • Lymphocytic and monocyte infiltration of the myelin sheath and perivascular region.
Clinical presentation • Typically, approximately 2 weeks after viral infection or immunization, muscle weakness of the distal lower extremities develops suddenly. In 65% of cases, the disease manifests itself within 3 weeks after the infectious disease • Subsequently, muscle weakness spreads upward to the muscles of the arms, torso, neck, and cranial muscles—symmetrical flaccid tetraparesis is formed. In some cases, only the lower extremities or cranial nerves are affected • Sensory disorders are minimal, pain, parasthesia, hypoalgesia or hyperalgesia in the distal extremities are possible • Paresis of facial muscles and bulbar disorders (bilateral paresis of the oropharyngeal muscles) often occur • Paralysis of the respiratory muscles (5–10 % of cases) • Decrease and then loss of deep tendon reflexes • Autonomic disorders •• Inappropriate secretion of ADH (urinary retention) •• Arrhythmias •• Fluctuations in blood pressure.
Special studies • Electromyography - a significant decrease in the amplitude of the muscle response when stimulating the distal peripheral nerve. Conduction of nerve impulses is slowed • Lumbar puncture. An increase in protein content, sometimes significant (>10 g/l), begins a week after the manifestation of the disease, with a maximum of 4–6 weeks.
Differential diagnosis • Intoxication leading to disruption of neuromuscular transmission •• Poisoning with heavy metals (lead, arsenic) •• Poisoning with industrial substances (acrylamide, carbon disulfide, trichlorethylene, rapeseed oil, organophosphorus compounds) •• Intoxication when taking drugs: gold preparations ... nerves in oncological diseases • Infectious diseases •• Acute poliomyelitis •• Diphtheria (complicated by paralysis) •• Botulism.
Treatment • According to indications - mechanical ventilation (in 10–23% of cases), tracheostomy • Intravenous administration of immunoglobulin (0.4 g/kg/day for 5 days) • In severe cases - plasmapheresis • Sufficient fluid intake to maintain diuresis at level 1 –1.5 l/day under the control of serum electrolyte concentrations • Physiotherapy to prevent contractures • Prednisolone up to 80–120 mg/day orally is prescribed only for chronic inflammatory dimyelinating polyradiculoneuropathy • Syndromic therapy • Heparin 5,000 units subcutaneously 2 times a day - for the entire period of bed rest.
Complications • During the disease: paralysis, respiratory failure and aspiration, arterial hypertension or hypotension, arrhythmias, urinary retention, depression • Consequences of the development of the chronic stage: chronic inflammatory demyelinating polyradiculoneuropathy.
Course and prognosis • Characterized by an acute onset and progressive course. Typically, the progression of the process continues for 2–4 weeks, then gradually, over about a year, functional restoration occurs • In 7–22% of patients, residual neurological deficits are detected (weakness, decreased reflexes) • In 10%, a relapse occurs within one year after recovery or severe complications develop • Mortality - about 3% • The prognosis for unclear etiology is favorable in approximately 50% of cases.
Synonyms • Guillain–Barré–Strol syndrome • Acute primary idiopathic polyradiculoneuritis • Infectious idiopathic polyneuropathy • Radiculanglionitis • Landry–Guillain–Barré syndrome • Acute demyelinating polyradiculoneuropathy Guillain–Barré
ICD-10 • G61.0 Guillain–Barré syndrome
Application. Miller Fisher syndrome is an atypical variant of Guillain–Barré syndrome, in which flaccid distal tetraparesis with areflexia is combined with bilateral external or total ophthalmoplegia and cerebellar ataxia.
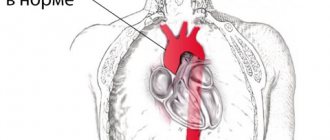
GEF in the ventricles of the fetal heart
The frequency of occurrence is about 7%. GEF in the ventricles of the fetal heart is a small formation inside the heart cavity, which, when performing ultrasound, is comparable in echostructure to bone tissue. The main ultrasound criteria for pathology include:
- Size from 1 to 6 mm;
- Location – on the papillary cardiac muscle or on the chordae tendineus;
- Most often detected in the left ventricle (94% of cases);
- Does not produce a specific sound shadow during ultrasound;
- Moves in sync with the heart valves.
Alternative names for GEF in the ventricles of the fetal heart are “golf ball”, “pea”. An ultrasound diagnostic doctor will not always be able to detect changes in the cavities of the fetal heart, but in any case, if a “pea” is detected, there is no need to panic. Possible outcomes of the GEF include:
- Reduction in size and disappearance during pregnancy (about 30%);
- Preservation throughout the entire period of gestation and absence of problems after the birth of the child (about 60%);
- Detection of changes in the heart of a newborn baby (3-5%), which do not require surgery and disappear on their own by the age of 4 years of the child’s life.
In rare cases, when GEF is combined with other signs of congenital anomalies, it can be expected that additional examination using invasive diagnostic methods will reveal chromosomal abnormalities or malformations in the child.
Goldenhar syndrome - symptoms and treatment
Goldenhar syndrome is a rare congenital anomaly that causes changes in the size and shape of facial structures. Usually the changes are localized on one side of the face, causing its asymmetry, but sometimes bilateral lesions occur.
This syndrome belongs to a spectrum of congenital anomalies of the skull and facial structures, which have the general term “craniofacial microsomia”. It refers to the reduction of any body structure within the craniofacial region.
Synonyms of the syndrome: oculoauricular dysplasia, facio-auriculo-vertebral association, syndrome of the 1st and 2nd branchial arches, otomandibular dysostosis, hemifacial microsomia, etc.
The approximate incidence of Goldenhar syndrome is 1 case in 3500-25000 newborns [9]. It occurs 2 times more often in boys than in girls.
The exact causes of the disease are not fully known today [1][2][3][4]. Most cases occur incidentally in families with no known medical history. However, 1-2% of patients with Goldenhar syndrome have close relatives with a similar disorder. This indicates the role of genetic factors in the occurrence of this pathology [4][5]. In particular, the involvement of the MYT1 gene, located at the q13.33 locus of chromosome 20, is assumed.
Another possible factor in the development of Goldenhar syndrome is chromosomal abnormalities - loss or duplication of a chromosome section. Typically, people with these disorders may have combined developmental defects such as abnormalities of the heart, lungs, kidneys, limbs and central nervous system [1][2][5][6].
Some researchers believe that the formation of the syndrome is facilitated by impaired blood flow or external damaging factors:
- taking certain medications that are contraindicated during pregnancy;
- bad habits;
- chemical and physical agents affecting the fetus at 3-8 weeks of intrauterine development [5][6].
It is also impossible to exclude the role of such obstetric and gynecological factors as previous abortions, diabetes mellitus and obesity [18].
The first descriptions of congenital anomalies of facial structures were found in ancient writings dating back to 2000 BC. e. In Colombia and Mexico, ancient ceramic items were found with images of various variants of hemifacial microsomia, including hereditary ones: one of the items depicted a parent holding a child in their arms, who had similar facial anomalies [10].
Possible reasons
Problems of intrauterine development most often arise against the background of the following factors:
- Intrauterine infection with viruses or bacteria;
- Chromosomal aberrations (calcium deposits on the walls of the heart cavities are common in children with Down syndrome);
- Congenital fibrous tissue dysplasia;
- Vascular disorders and fetal heart dysfunction;
- Immunological disorders due to underdevelopment of the thymus;
- Anomalies of heart development in a baby.
GEF in the area of the heart in a baby is serious: if there are risk factors, the doctor will prescribe an additional examination to exclude a dangerous congenital pathology in the baby.
After identifying a hyperechoic focus in the fetal heart, you should be observed by a doctor and do an ultrasound as prescribed by the doctor
Medical Internet conferences
This syndrome was first described by the American neurosurgeon Walter Dandy in 1921 and Earl Walker in 1944. Among live-born children, the incidence rate is from 1:5000 to 1:25000, and among children with congenital hydrocephalus it ranges from 3.5 to 12% [2] .
Dandy-Walker Syndrome is a malformation of the brain (cerebellum and surrounding cerebrospinal fluid spaces), which is characterized by a triad of symptoms: hypotrophy of the cerebellar vermis and/or cerebellar hemispheres, cysts of the posterior cranial fossa, hydrocephalus of varying degrees. This syndrome according to ICD X is coded in the category of developmental anomalies under the code Q 03.1, as atresia of the foramina of Magendie and Luschka [3, 4].
According to modern concepts, the etiology of Dandy-Walker syndrome is extremely heterogeneous, since various factors are involved in its occurrence: hereditary (chromosomal and genetic) and exogenous teratogens; it is assumed that factors such as viral infection (CMV, rubella) also play a role; alcohol intake, diabetes during pregnancy. In 1/3 – 1/2 of cases, Dandy-Walker syndrome is combined with other various congenital syndromes (Table 1) [2].
In 70% of cases, the defect is combined with other brain anomalies - agenesis of the corpus callosum, encephalocele, polymicrogyria, agyria, heterotopia of gray matter, as well as lesions of other organs and systems (polydactyly, syndactyly, congenital heart defects, polycystic kidney disease, cleft palate and etc.) [5].
Among the main hypotheses for the occurrence of Dandy-Walker syndrome are the following:
— stopping embryonic development in the process of formation of the rhombencephalon;
— atresia of the outlet from the IV ventricle with delayed opening of the foramen of Magendie;
- the appearance of the choroid plexus of the fourth ventricle in the middle of the thin roof of the rhombencephalon.
Prenatal functional diagnostics specialists distinguish complete and incomplete, as well as closed and open forms of Dandy-Walker syndrome. The full form is characterized by agenesis of the cerebellar vermis and the presence of obvious communication between the fourth ventricle and the cyst in the region of the cistern magna. The incomplete form is partial agenesis of the lower part of the cerebellar vermis, and therefore, communication of the fourth ventricle with the cistern magna cyst is not traced throughout the entire length of the vermis. The open and closed forms differ in the presence or absence of occlusion of the foramina of Luschka and Magendie and the connection of the ventricle with the subarachnoid space (Fig. 1, 2) [6].
Dandy-Walker syndrome is characterized by pronounced clinical polymorphism: from almost normal postnatal development to severe disability and even death of the child. Perinatal outcomes largely depend on the depth of damage to the central nervous system (progressive hydrocephalus), as well as on the presence of concomitant pathology, which accompanies this syndrome in 60–75% of cases [2, 5].
The prognosis for life and health with Dandy-Walker syndrome depends on the presence of combined developmental anomalies, chromosomal abnormalities and the timing of diagnosis. According to the literature, postnatal morbidity and mortality rates are higher in cases where the syndrome is diagnosed prenatally rather than postnatally [2].
Treatment of this disease is symptomatic. If there are signs of increasing intracranial hypertension, shunt surgery is performed. The outcome is often fatal, in 90% of cases in the first years of life [7].
Under observation was a full-term newborn boy L., born from the 3rd pregnancy, which occurred against the background of late registration (at 30 weeks). From 35 weeks of gestation, polyhydramnios was noted and for the first time, an ultrasound examination revealed an anomaly of fetal development - Dandy-Walker malformation.
Prenatal ultrasound picture at 35 weeks of gestation: the shape of the head is abnormal (strawberry-shaped). The bones are not deformed when pressed by the sensor. Expansion of the lateral ventricles, anterior horns up to 10 mm on the right and left. The cavity of the transparent septum is up to 9.9 mm. Rear horns – 11 mm, left – 12 mm. Enlargement of the IV ventricle up to 12 mm was noted. There is hypoplasia of the cerebellar vermis, expansion of the cisterna magna up to 12 mm.
A child from the third term birth is in cephalic presentation. Amniotic fluid is light yellow in a volume of two liters. The child's body weight at birth was 2890 g, height - 49 cm, head circumference - 35 cm, chest circumference - 33 cm. Apgar score 4 - 7 points.
The child’s condition after birth was severe; he was admitted to the NICU from the delivery room due to stage III respiratory failure and severe neurological symptoms. From the moment of admission he was on mechanical ventilation, from the third day of life he was transferred to respiratory support in the CPAP mode, and from the 7th day of life to spontaneous breathing. Oxygen therapy was carried out according to indications. Enteral nutrition began from the first day in the trophic volume with subsequent expansion, assimilation.
Neurologically: the child is forced to lie on his side with his head thrown back, the cry is monotonous, there are floating movements of the eyeballs, the “setting sun” symptom, unstable horizontal nystagmus. Irregular shape of the skull, enlarged brain part, increase in head circumference over 2 weeks was 2.5 cm. There was a divergence of the skull bones, the sutures were open up to 0.5 cm, with an increase in the size of the large fontanel to 5 × 8 cm. The pupils were medium magnitude, the photoreaction is preserved. Spontaneous motor activity and muscle tone are reduced. Dystonia in the limbs. Physiological reflexes are depressed, tendon reflexes are reduced, and symmetrical.
Noteworthy was the presence of bilateral clubfoot with varus deformation of both feet, hands and phalanges, hypertelorism, saddle-shaped bridge of the nose, overhanging forehead and back of the head, wide tongue, short neck (Fig. 3).
On the 14th day of life, after the condition had stabilized, the child was transferred to the second stage of treatment, to the neonatal pathology department for further observation and treatment.
Neurosonography picture in the first day of life: in the posterior cranial fossa, coronal and sagittal scans show a large anechoic (“cyst”) formation, including dilated III and IV ventricles; the cerebellar hemispheres are sharply reduced, the vermis is not defined; the cerebellar tentorium is displaced upward.
In the dynamics, according to the NSG, periventricular ischemia was revealed, Dandy-Walker syndrome, ventriculomegaly (as part of a symptom complex) and increased cerebral vascular resistance were confirmed.
The child was examined by a neurologist, ophthalmologist, and geneticist, and a diagnosis was made: Congenital malformation of the central nervous system - Dandy Walker syndrome. Hypoxic-ischemic damage to the central nervous system. After an examination by a neurosurgeon, a diagnosis was made: congenital malformation of the central nervous system, Dandy-Walker syndrome, hydrocephalic syndrome.
At the time of examination, he did not require neurosurgical treatment; recommendations for care and treatment were given.
According to the results of DEHO-CG, a congenital heart defect was revealed: combined pulmonary artery stenosis (valvular-subvalvular), ASD with left-to-right shunting. Open oval window with a diameter of 2.0 mm.
The child was consulted by a cardiologist and cardiac surgeon, and recommendations were given.
Ultrasound of the abdominal cavity and kidneys revealed no pathology.
After examination by an orthopedic surgeon, congenital bilateral clubfoot was confirmed.
Due to the stabilization of the child’s condition, after examination and treatment, as well as the mother’s refusal of parental rights, the boy was transferred to an orphanage in the city of Marx on the 57th day of life.
In conclusion, it should be said that this observation is of great interest from a clinical point of view, since it does not occur so often in the daily practice of a doctor.
Early diagnosis of complex genetic syndromes, which include the described clinical observation, presents certain difficulties. In my opinion, in such situations, it is justified to make a syndromic diagnosis with clarification of developmental anomalies based on an analysis of the totality of clinical data and additional examination methods. An accurate nosological diagnosis is important not only for genetic analysis and medical genetic counseling, but also, above all, for prevention and treatment. Without a reliable clinical diagnosis, analysis of factors and their theoretical understanding are impossible.
GEF in the ventricles of the fetal heart: medical tactics
In most cases, if a “golf ball” is detected in the area of the child’s heart, it is necessary to continue observation: a control ultrasound examination during pregnancy should be performed by a certified diagnostician using a modern 3D device. If there is a suspicion of chromosomal abnormalities in the fetus, one must agree to invasive research methods.
After the birth of a child, it is important in the first days and during the first years of the baby’s life to be regularly examined by a pediatrician and cardiologist.
Detection of any changes in the baby’s heart is a significant reason for a careful assessment of the child’s intrauterine development.
Hirschsprung's disease in children: no need to panic
Many genetically determined diseases are incurable, but there are lucky exceptions. For example, Hirschsprung's disease in children. Mikhail Yuryevich Kozlov, head of the surgical department of the Morozov Children’s Hospital, coloproctologist, pediatric surgeon of the highest qualification category, candidate of medical sciences, holder of the “Moscow Doctor” status, spoke about how this disease manifests itself, why it is dangerous and how it should be treated.
What do parents need to know about Hirschsprung's disease?
This is one of the most severe congenital defects of the colon, but it is curable. At certain stages of embryonic development, the wall of the colon does not receive a normal structure, that is, nerve ganglia (clusters of nerve cells) are not formed in it. As a result, the intestines do not perform their normal functions. The cause of the disease is a genetic failure. Changes in chromosomes can be either hereditary or spontaneous. Fortunately, the disease is quite rare: 1 case in 5,000 newborns. Today, the likelihood of having a child with Hirschsprung's disease can be determined using a genetic test. This is especially true if there has already been a case of this disease in the family.
What symptoms suggest Hirschsprung's disease?
The absence of nerve ganglia (or their deficiency) leads to the fact that the intestinal wall does not stop - it cannot push feces towards the exit. A condition arises that is popularly called constipation, and we, doctors, talk about stool retention. This is the main manifestation of Hirschsprung's disease. However, everything is individual: if for one child going to the toilet 3 times a day is considered the norm, then for another, stool 2 times a week will not be a deviation. Provided that such stool retention is not combined with other clinical manifestations. But progressive constipation, along with a lack of appetite, bloating and developmental delays in a child, is already a reason to immediately consult a doctor to rule out Hirschsprung’s disease at an early stage.
What does Hirschsprung's disease lead to if left untreated?
The consequences of the disease are extremely severe. If the child is not operated on in time, he will stop going to the toilet, a large amount of feces will accumulate and his colon will overstretch. As a result, chronic fecal intoxication will occur: the stomach will begin to swell, appetite will disappear, and vomiting will appear. If a child has been suffering from this problem for a long time, then, against the background of intoxication, by the age of 3–4 years of life he will develop encephalopathy with all the ensuing consequences: developmental delay, uncontrolled psycho-emotional behavior, problems with attention, etc.
Is Hirschsprung's disease currently curable?
Yes, 100%, but we are talking exclusively about surgical treatment. Hirschsprung's disease is not a case where some mythical drops will help; it can only be treated by surgeons. The operation is called "single-stage laparoscopic colon resection." For the first time in Russia, we performed this operation at the Izmailovo Children's Hospital back in 2004, and at the Morozov Hospital we have already continued to improve this technique. Since then, we have successfully operated on more than 700 children, and our hospital is one of the most advanced in terms of treating this disease.
The essence of the operation is extremely simple: laparoscopically, that is, through three small punctures on the anterior abdominal wall, the aganglionic (i.e. non-functional) section of the colon is excised, its healthy part is lowered down and sutured, roughly speaking, to the butt. Then a direct anastomosis is performed, without any stoma (an artificial opening between the intestinal cavity and the environment). The surgical intervention is performed under general anesthesia with the obligatory addition of epidural anesthesia in order to minimize the use of narcotic analgesics in the child in the immediate postoperative period. On average, a small patient stays in the hospital for no more than two weeks.
All parents are concerned about the question of how safe such an operation is for the child, since during it about 40 cm of the colon can be removed?
Theoretically, any operation, even banal removal of a wart, can have complications for the body. As for one-stage laparoscopic colon resection, over 16 years of experience in our clinic using modern equipment, we have perfected the surgical technique. Therefore, the risk of postoperative complications is 0.5-1%. Judge for yourself: if at the stage of mastering the technique the operation lasted 3–4 hours, today it takes no more than 40 minutes. In this case, two medical teams work simultaneously: one from the abdominal cavity performs the laparoscopic stage, the second from the perineum performs the perineal stage.
After laparoscopic surgery, is Hirschsprung's disease cured forever or will the child need some more treatment throughout his life?
Yes, after a competently performed laparoscopic resection of the colon in one stage, the child no longer needs any surgical intervention and leads a normal life. The only exceptions are secondary patients. Our colleagues in the regions are mastering this technique, but the level of equipment or experience does not always allow it to be performed flawlessly. Recently, we have had a sharp increase in the number of repeat operations: children are being admitted who have already been operated on 2-3 times. But repeated reconstructive surgical interventions are more dangerous in terms of the number of complications and the postoperative result is no longer so ideal.
Can you share the most interesting case from your practice related to Hirschsprung's disease in a patient?
For the operating surgeon, every case is interesting, and it cannot be otherwise. But the most memorable is, of course, the very first patient. For me, this was a boy named Ilya, who was successfully operated on in 2004. Now he is already an adult young man who is preparing to enter a university. He is absolutely healthy and leads a full life. Therefore, I want to give only one piece of advice to all parents: don’t panic! You just need to operate on the child as soon as this disease is detected. Diagnosed with Hirschsprung's disease in a month? This means that we need to operate within a month. If diagnosed in a year, it is necessary to operate in a year. Don't wait and don't be afraid.
Can only Muscovites undergo surgery at the Morozov Hospital in Moscow, or is it also available for children from other regions of Russia?
In our hospital, this operation is performed free of charge for all residents of Russia under the compulsory medical insurance policy! The main thing is to contact us for help in a timely manner.
Source: Moscow - the capital of health