Pheochromocytoma
− a tumor of chromaffin tissue that produces biologically active substances (adrenaline, norepinephrine, dopamine). In 90% of cases, pheochromocytomas arise in the adrenal medulla, in 8% in the aortic lumbar paraganglia. Much less often, tumors are localized outside the adrenal glands: in less than 2% of cases - in the abdominal and chest cavity and in less than 0.1% of cases - in the neck. Almost 50% of patients with pheochromocytoma have arterial hypertension, and 40-50% experience hypertensive crises. In patients with pheochromocytoma, blood pressure is unstable, fluctuates widely and is poorly controlled with medications.
Pathogenesis
One of the factors influencing blood pressure fluctuations in pheochromocytoma is a significant depot of unclaimed catecholamines in the endings of the sympathetic nerves. Any stimulation of the sympathetic system can provoke a crisis caused by the neurogenic effects of norepinephrine released from the synaptic store rather than from the chromaffin tumor. In this case, there will be no significant increase in catecholamine levels. The clinical manifestations of pheochromocytoma are extremely varied, which is explained by many reasons:
- variability in the development of the receptor apparatus for catecholamines in different organs;
- excessive deposition of norepinephrine due to the mechanism of reverse neuronal uptake in the presynaptic endings of the sympathetic nervous system;
- a disorder of receptor sensitivity against the background of long-standing hypercatecholaminemia;
- violation of general systemic control over hemodynamics in conditions of relative hypovolemia occurring in patients with pheochromocytoma;
- intratumoral methylation of adrenaline, norepinephrine and dopamine, which leads to the formation of inactive fractions of catecholamines (metanephrine, normetanephrine and methoxytyramine)
Among the reasons influencing the decrease in the volume of circulating fluid in patients with pheochromocytomas, centralization of blood circulation is noted. This occurs due to increased peripheral vascular resistance and the shunting effect. A significant component in the formation of hypovolemia is the release of fluid from the vascular bed into the third space. This occurs as a result of changes in the permeability of the vascular wall due to persistent vasoconstriction and due to the formation of fibromuscular dysplasia during prolonged vascular spasm. Important points that influence the occurrence of hypovolemia are increased sweating and chronic constipation.
Hypovolemia in pheochromocytoma is one of the leading syndromes that determine the severity of the patient's condition. Having a masking effect on the results of measuring peripheral blood pressure, which often leads to diagnostic errors and incorrect treatment decisions, hypovolemia syndrome is largely responsible for microcirculation disorders in vital organs.
In the pathogenesis of changes in the cardiovascular system, the occurrence of morphological changes in the myocardium against the background of hypercatecholaminemia plays an important role. One of the most common misconceptions that leads to late detection of pheochromocytoma is overdiagnosis of myocardial ischemia. It should be noted that both electrocardiographic and laboratory signs of myocardiocyte destruction are nonspecific.
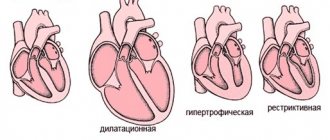
The occurrence of arrhythmias, myocardial necrosis, laboratory cytolytic syndrome and electro- and echocardiographic changes in the vast majority of cases of pheochromocytoma is not associated with changes in the coronary circulation. The main cause of cardiotoxic changes in hypercatecholaminemia is an intracellular disruption of the action of enzymes responsible for the phosphorylation process. This leads to changes in inter- and intracellular ion exchange and the oxidative intracellular cycle (the so-called toxic catecholamine myocardial dystrophy). High afterload against the background of non-coronary myocardial dystrophy or myocardial necrosis can lead to acute left ventricular failure (cardiac asthma, pulmonary edema). With a long history of pheochromocytoma and progressive cardiosclerosis against the background of myocardial hypertrophy, concentric and then dilated cardiomyopathy occurs, inevitably leading to CHF. Paroxysms of cardiac arrhythmia are a high risk factor for sudden cardiac death in these patients.
Catecholamine shock is the most dangerous manifestation of pheochromocytoma. With its development, persistent uncontrollable hypotension is noted against the background of cardiac arrhythmias, which lead to ineffective cardiac output. This is due, on the one hand, to a change in the sensitivity of adrenergic receptors against the background of constant intense stimulation, depletion of cells of the conduction system of the heart, and on the other hand, to a change in the mechanisms of inactivation of catecholamines.
An important factor in increasing hypotension is hypovolemia, which is directly proportional to the intensity and duration of catecholamine intoxication. With catecholamine shock, a paradoxical situation arises when a vasoconstrictor status is noted in the central vessels (systolic blood pressure in the aorta at a level of 300 mm Hg or more) and vascular hypotension in the periphery. Treatment measures carried out under these conditions do not contribute either to the improvement of the patient’s condition or to the correct diagnosis.
Of the clinically significant pathophysiological effects in pheochromocytoma, it is necessary to pay attention to the occurrence of secondary diabetes or impaired glucose tolerance, which is due to the acceleration of glycogenolysis in the liver, a decrease in insulin production due to stimulation of α-adrenergic receptors of the pancreas.
General information
Pheochromocytoma (chromaffinoma) is a benign or malignant hormonally active tumor of chromaffin cells of the sympathetic-adrenal system, capable of producing peptides and biogenic amines, including norepinephrine, adrenaline, and dopamine. In 90% of cases, pheochromocytoma develops in the adrenal medulla; in 8% of patients it is localized in the area of the aortic lumbar paraganglion; in 2% of cases - in the chest or abdominal cavity, in the pelvis; extremely rarely (less than 0.1%) - in the head and neck area.
Clinical endocrinology describes pheochromocytomas with intrapericardial and myocardial localization, with a predominant location in the left side of the heart. Pheochromocytoma is usually detected in people of both sexes aged 20-40 years; in children it is more common among boys (60% of observations). Pheochromocytoma is a common cause of arterial hypertension and is detected in approximately 1% of cases in patients with persistently elevated diastolic blood pressure.
Malignant pheochromocytomas account for less than 10% of cases, they are usually extra-adrenal and produce dopamine. Metastasis of malignant pheochromocytomas occurs in regional lymph nodes, muscles, bones, liver and lungs.
Pheochromocytoma
Clinical picture
The main symptom is arterial hypertension (constant, paroxysmal or mixed form). A characteristic feature of the hemodynamic (hypertensive) crisis in pheochromocytoma is its short duration and so-called self-limitation.
Additional symptoms include:
- orthostatic hypotension;
- sweating;
- constant headaches;
- feeling of inner trembling, anxiety;
- general weakness, decreased ability to work.
The classic course of pheochromocytoma is indicated when patients have hypertensive crises with a sudden increase, primarily in systolic blood pressure, which can reach 300 mm Hg. Crises can be provoked by slight physical activity, palpation of the abdomen, sometimes by taking β-blockers, and when pheochromocytoma is localized in the wall of the bladder, by urination; are accompanied by rapid heart rate (up to 180/min), arrhythmias and/or changes in the electrocardiogram (ECG) like acute coronary ischemia (usually not associated with coronary circulation disorders, but due to the direct toxic effect of catecholamines).
Also during an attack, tremors, tinnitus, anxiety or fear, dilated pupils, sweating, chest or abdominal pain, nausea or vomiting are often noted. Vasoconstriction of the extremities under the influence of catecholamines can cause pain and paresthesia, intermittent claudication, Raynaud's syndrome, ischemia, and trophic ulcers. Hyperglycemia and glycosuria, leukocytosis may be detected. The duration of an attack can be from several minutes (usually) to several hours (much less often). The attack usually ends suddenly. With pheochromocytoma, decompensation of previously undetected diabetes mellitus or impaired glucose tolerance is possible. When examining the fundus, spastic angiopathy is revealed. In pheochromocytoma, the severity of changes in the fundus does not correspond to the malignancy of the course of hypertension.
In approximately 10% of cases, pheochromocytoma is a familial disease and is inherited in an autosomal dominant manner. Pheochromocytomas and accompanying tumors of the thyroid gland and nervous tissue are of neuroectodermal origin, as evidenced by the presence of neuron-specific enolase in all these tumors. Apparently, the occurrence of such tumors is due to disturbances in the proliferation and differentiation of neural crest cells.
MEN type IIa. This hereditary syndrome is caused by a defect in one of the loci of chromosome 10. Components of the syndrome: medullary thyroid cancer, hyperplasia or adenoma of the parathyroid glands (clinically manifested as hyperparathyroidism), pheochromocytoma and (less commonly) bilateral adrenal hyperplasia.
MEN type IIb. EC components of the syndrome: pheochromocytoma, medullary thyroid cancer, mucosal neuromas, thickening of the corneal nerves, gastrointestinal ganglioneuromas; often Marfan-like appearance.
Other associated syndromes. 5% of patients with pheochromocytoma have neurofibromatosis (Recklinghausen's disease). A combination of pheochromocytoma, neurofibromatosis and somatostatin-containing carcinoid tumor of the duodenum has been described. A combination of pheochromocytoma with Hippel-Lindau disease (retinocerebellar hemangioblastomatosis) and acromegaly was observed.
Causes
This disease most often occurs in people aged 25 to 50 years. The development of this type of tumor is mainly observed in middle-aged women. If we talk about children, this diagnosis is made more often in boys. But in general, pheochromocytoma tumors are a relatively rare phenomenon. Moreover, approximately 10% of the total number of such problems are related to the family form. This means that this type of tumor was present in at least one of the patient’s parents. Continuing to consider the causes of this type of disease, it is worth noting that it may be one of the components of the syndrome of multiple endocrine tumors.
Diagnostics
The regulation of catecholamine secretion does not obey the “feedback” law and directly depends on external and/or internal stress factors. Diagnosis is complicated by the extremely short half-life of adrenaline and norepinephrine, as well as the pulse increase of these hormones in response to any stress in healthy people.
General clinical studies
Routine studies are indicative in nature, but have some significance in the diagnosis of pheochromocytoma. There is a proven relationship between the release of catecholamines and an increase in glycemia and an increase in leukocytosis in the peripheral blood. It should be noted that these indicators should only be determined at the time of crisis or immediately after it. This becomes possible only in a hospital setting and during a crisis course of pheochromocytoma, which significantly reduces the diagnostic value of this technique.
Hormonal laboratory tests
Determining the level of catecholamines in the blood for diagnosing pheochromocytoma is not very informative due to their rapid biological degradation and wide range of variation. Reference values for adrenaline range from 0 to 110 pg/ml (pg/ml x 5.46 = pmol/l) and for norepinephrine 70-750 pg/ml (pg/ml x 5.91 = pmol/l). The greatest diagnostic value is determining the level of intermediate products of catecholamine metabolism, which remain in the bloodstream for a long time, which makes it possible to lengthen the period from the moment of crisis to taking blood samples. Free metanephrines (metanephrine and normetanephrine) in plasma and conjugated metanephrines in urine have diagnostic value. Plasma metanephrines are determined in free form. The level of methylated catecholamine derivatives (MCD) is an integrative indicator of tumor activity over 24 hours. The method for determining free MIC in plasma has high sensitivity and specificity and reaches 95–100%. If the synthesis of adrenaline predominates in the tumor, then there is an increase in the level of metanephrine, if norepinephrine - then normetanephrine, dopamine - 3-methoxytyramine, with a mixed type of tumor production, an increase in all of these indicators is possible. A slight excess of the upper limit of plasma fractionated metanephrines, 3-methoxytyramine is associated with a low probability of the presence of a tumor, a fourfold excess is associated with an almost 100% probability of a mass formation. Reference values for metanephrines in plasma are up to 90 ng/ml, and for normetanephrine - up to 200 ng/ml. Simultaneous testing of catecholamine metabolites in plasma and urine significantly increases the specificity of the study, reducing the likelihood of false-positive results. When examining urine, the level of metanephrines in an adult should not exceed 320 mcg/day (mcg/day x 5.07 = nmol/day), and for normetanephrines - up to 390 mcg/day (mcg/day x 5.46 = nmol/day ). When performing these tests, the conditions of the preanalytical stage must be strictly observed. Before studying the level of catecholamines, it is necessary to exclude bananas, pineapples, cheese, strong tea and coffee, and products containing vanillin from the diet. A few days before the study, stop taking tetracycline antibiotics, quinidine, reserpine, tranquilizers, adrenergic blockers, and MAO inhibitors. The study is carried out on an empty stomach. Before blood is drawn, the patient is given complete physical and emotional rest for at least 20 minutes.
Topical diagnosis of pheochromocytoma
Methods of radiological diagnosis of the adrenal glands are the main type of topical diagnosis. In the case of an adrenal tumor, the information content of CT is from 85% to 95%. When conducting an MRI study, a lipid-rich tumor is quite clearly visualized on an MRI study, which makes it possible to accurately make this diagnosis using this method. In this case, the greatest information content is achieved, which is almost 100%. The greatest difficulties arise in the diagnosis of extra-adrenal pheochromocytomas, which occur in 10% of cases. If the tumor location is unclear, scintigraphy is performed with metaiodobenzylguanidine (MYBG), which has maximum affinity for tumor cells. MyBG is concentrated in the cells of chromaffin tissue, while normal adrenal tissue rarely absorbs the isotope, but up to 90% of pheochromocytes absorb it. In special cases, with an obvious clinical picture confirmed by laboratory tests and the absence of visualization on CT/MRI, a PET scan with fluorodeoxyglucose labeled with an isotope with a short half-life is performed. This method is the final one in the topical diagnosis of pheochromocytoma.
Treatment
Drug treatment
The main goal of drug therapy is preparation for surgical treatment. The strategic goal of preoperative preparation is to increase functional reserves. The main drug of choice for the preoperative preparation of patients with pheochromocytoma at the present stage is the selective prolonged α1-blocker doxazosin . The drug is used orally; it affects the entire spectrum of α1-adrenergic receptors of resistive vessels. Half-life is up to 22 hours. The effective dose is achieved after 2–3 hours. Prescribed at a dose of 1–16 mg per day for 1–2 doses with an initial dose of 2–4 mg/day. In the absence of hypovolemia, the drug does not cause hypotension in the interictal period. Doxazosin is dosed according to the hypotensive effect and the disappearance of hypovolemic manifestations.
Unlike α-blockers with a symptomatic mechanism of action, the use of α-methylparatyrosine is based on the pathogenetic aspects of pheochromocytoma. The drug blocks tyrosine kinase, which regulates the synthesis of catecholamines. The drug is prescribed at an initial dose of 250 mg 4 times a day, followed by a possible increase to 4 g/day.
Among the side effects, it is necessary to note the possibility of prolonged intra- and postoperative hypotension, as well as a different range of psychasthenic manifestations, and therefore its use as the main means of preoperative preparation is limited. α-Methylparatyrosine is considered the drug of choice for the treatment of metastatic pheochromocytoma.
In combination with antihypertensive drugs, one of the main stages of preoperative preparation may be to achieve a β-adrenergic blocking effect in case of tachycardia or rhythm disturbances. Given the existing abundance of drugs in this group, preference is given to cardioselective drugs. This is important because of the undesirable effects of β2-adrenergic receptor blocking: hypokalemia and paradoxical hypertension. Prescription of β-blockers is possible after achieving an α-blocking effect. If this condition is not met, a paradoxical worsening of the course of crisis hypertension cannot be ruled out, which is associated with the leveling of the β2-dilating effect of adrenaline. Before surgery, long-acting α-blockers and drugs that affect the synthesis of catecholamines must be discontinued.
Surgery
The main method of treatment with chromaffin is surgical removal of the tumor. At the present stage, it is most preferable to use α-blockers (phentolamine, trodiphene) or peripheral vasodilators (sodium nitroprusside) for effective control of intraoperative hemodynamics. The use of sodium nitroprusside has provided anesthesiologists with a powerful, rapidly titrated drug for the management of sudden fluctuations in blood pressure. The α-blockers used are not as easily and readily titrated (dosed) as sodium nitroprusside. Its half-life is about 1 minute, the same figure for phentolamine (tropafene) is 19 minutes. It is obvious that the aftereffect after the administration of α-blockers can aggravate the hypotension that occurs after disconnection of the catecholamine-producing tumor from the central bloodstream.
Heart rhythm disturbances (tachycardia, supraventricular extrasystoles) are treated with β-blockers. The optimal agent, combining cardioselectivity and a short period of action, which is important when prescribing it intraoperatively, according to most authors, is esmolol. Removal of pheochromocytoma, like no other operation, requires clear interaction between the surgeon and anesthesiologist, since changes in blood pressure directly depend on their actions. Adrenal tissue preservation techniques preserve cortical function; however, the alternative may be local tumor recurrence. Surgical treatment in conditions of a scar process carries a greater likelihood of complications than during primary operations. The gold standard for the extent of surgical intervention for pheochromocytoma is adrenalectomy with tumor.
How to treat pheochromocytoma
To treat pheochromocytoma, surgical techniques are used primarily. Before the operation, the main symptoms of the crisis are relieved with the help of medications and the condition of the patients is stabilized.
Attacks are relieved by lowering blood pressure and heart rate. A combination of α-blockers (phenoxybenzamine, tropafen, phentolamine) and β-blockers (propranolol, metoprolol) is used. Also, in case of a crisis increase in blood pressure, phentolamine and sodium nitroprusside are administered.
Surgery for pheochromocytoma is performed exclusively by laparotomy. There is always a high risk that other tumors or metastases located outside the adrenal glands will be detected in the patient’s body. During the operation, special attention is paid to monitoring blood pressure (peripheral and central). The usual treatment for pheochromocytoma is a complete adrenalectomy. If the pheochromocytoma tumor is one of the manifestations of multiple endocrine neoplasia, the adrenal glands are removed on both sides to avoid further spread of the tumors.
After surgery to remove a pheochromocytoma, your blood pressure drops. When this does not happen, they look for another focus located outside the adrenal tissue.
If pheochromocytoma is detected in a woman carrying a child, blood pressure is first stabilized. Then a decision is made to terminate the pregnancy or deliver by caesarean section. Only after this an operation is performed to remove the tumor.
The malignant form of pheochromocytoma with multiple metastases requires chemotherapy using cyclophosphamide, vincristine, dacarbazine and other drugs.