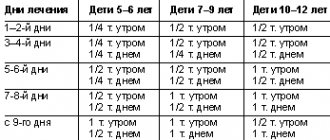
Classification and epidemiology
In AS, the myocardium is infiltrated with an insoluble glycoprotein, amyloid, formed from various serum or locally produced precursor proteins. These include constant proteins that are independent of amyloid type, such as serum amyloid P protein (SAP), but about 80% of the amyloid mass consists of proteins that differ between amyloid types. Modern classification of amyloidosis is based on differences in amyloidogenic precursor proteins. About 30 such proteins are currently known; Accordingly, there are about 30 forms of amyloidosis. Each form is designated by an abbreviation that includes the designation of the precursor protein: AL (L - light chains of immunoglobulins), ATTR (TTR - transthyretin), CAA (serum amyloid A), etc. (see table). According to this classification, primary and myeloma-associated amyloidosis should be abbreviated AL, since amyloid in these forms is formed by light chains of immunoglobulins. In systemic forms of amyloidosis (most cases of AL, as well as ATTR, AA, etc.), the amyloidogenic precursor protein circulates in the blood and can be deposited in various tissues, the tropism for which differs among different proteins. In addition, local amyloidosis, for example in the atria, may develop due to local production of the amyloidogenic variant of atrial natriuretic factor (AANF).
. Classification of cardiac amyloidosis.
The most common variant of systemic amyloidosis is AL amyloidosis, one of the many forms of plasma cell dyscrasia. The term “plasma cell dyscrasia” unites a group of conditions caused by pathology of plasma cells in the bone marrow, and less commonly, other tissues. In this case, in the bone marrow there is a dominance of the pathological clone of plasma cells that synthesize abnormal immunoglobulins or their components: heavy or light chains. In addition to AL amyloidosis, plasma cell dyscrasias include multiple myeloma, Waldenström macroglobulinemia, etc.
In AL amyloidosis, a pathological clone of plasma cells synthesizes light chains of immunoglobulins, the deposition of which in tissues is accompanied by the formation of insoluble amyloid fibrils. AL amyloidosis is the most severe, generalized form of the disease, affecting many tissues. The heart is affected in 90% of patients, half of whom develop diastolic heart failure (HF) [2]. Death usually occurs from heart failure or arrhythmias [4].
The isolated cardiopathic variant of AL amyloidosis is rarely diagnosed - in less than 5% of patients [5]. Isolated cardiac damage is characteristic of AANF amyloidosis, in which infiltration of the atria with amyloid formed from atrial natriuretic peptide is noted. This form of amyloidosis is found in more than 95% of people over 80 years of age [6]. AANF amyloidosis rarely leads to HF [2], but can cause the development of supraventricular arrhythmias [7–9].
Heart damage is typical for acquired and various variants of hereditary ATTR amyloidosis. This form is caused by the deposition of the protein that transports thyroid hormones and retinol - transthyretin. It is secreted by the liver in a tetrameric form consisting of four transthyretin molecules. With age, a decrease in the activity of hepatocyte enzyme systems leads to an increase in the secretion of amyloidogenic monomeric forms of transthyretin and, as a result, the development of amyloidosis. Acquired ATTR amyloidosis is usually called systemic senile amyloidosis (affects up to 25–36% of people over 80 years of age). Amyloid is found in many organs, but the most significant deposits are found in the heart. Typically, the disease has a benign, asymptomatic course, manifested by blockade of the anterior branch of the left bundle branch and slight thickening of the walls of the left ventricle (LV). With significant amyloid deposition, cardiomegaly and slowly progressive heart failure may develop [3].
In the presence of mutations in the transthyretin molecule, the assembly of tetramers in the hepatocyte is initially disrupted, which leads to the development of hereditary ATTR amyloidosis at an earlier age. Currently, about 100 point mutations and deletions of the transthyretin gene are known, 87 of which are amyloidogenic [10]. The disease is characterized by autosomal dominant inheritance, but the penetrance of the mutant gene varies widely. There are neuropathic, cardiopathic and ophthalmoleptomeningeal forms of the disease. Most often, the neuropathic form develops, which is characterized by progressive damage to the peripheral and autonomic nervous system. The predominant damage to one or another organ is largely due to the nature of the mutation. Thus, when isoleucine at position 122 is replaced by valine, senile amyloid heart disease develops without neurological manifestations; this mutation is found predominantly in African Americans [11]. Mutations that cause serious heart damage include the substitution of methionine for valine at position 30 (Portuguese variant of familial amyloidosis), the substitution of serine for isoleucine at position 84, and the substitution of alanine for threonine at position 60. Amyloid cardiopathy in hereditary ATTR amyloidosis is less severe, than in AL amyloidosis, but still leads to severe heart failure. In one of the most common types of amyloidosis, secondary AA amyloidosis, clinically significant cardiac damage is rarely detected - in less than 10% of cases [13]. Although AA amyloidosis is systemic in nature, and upon morphological examination amyloid deposits are found in various tissues, in this form of the disease clinical significance is mainly in the kidneys, and to a lesser extent in the liver, spleen and gastrointestinal tract.
Forms of the disease
Depending on the reasons that caused it, amyloidosis is divided into several clinical forms:
- senile (senile);
- hereditary (genetic, family);
- secondary (acquired, reactive);
- idiopathic (primary).
Depending on which organ amyloid deposits are predominantly deposited, the following are distinguished:
- amyloidosis of the kidneys (nephrotic form);
- cardiac amyloidosis (cardiopathic form);
- amyloidosis of the nervous system (neuropathic form);
- liver amyloidosis (hepapathic form);
- adrenal amyloidosis (epinephropathic form);
- ARUD-amyloidosis (amyloidosis of organs of the neuroendocrine system);
- mixed amyloidosis.
Kidney amyloidosis
Amyloidosis can also be local or systemic. With local amyloidosis, there is a predominant lesion of one organ, with systemic amyloidosis - two or more.
Morphology and pathogenesis
Amyloid deposition leads to significant concentric thickening of the myocardium, and dilatation of the ventricular cavities is not typical for AS. Amyloid infiltration of the myocardium is accompanied by a deterioration in its mechanical properties and leads to a significant decrease in contractility in combination with severe restrictive disorders of diastolic function. Modern research methods have shown that thickening of the myocardial walls in AS does not always correlate with impaired systolic and diastolic functions. Conventionally, three subtypes of AS can be distinguished: in patients with AL, hereditary ATTR and acquired senile ATTR amyloidosis [16].
In all cases, amyloid is found in the myocardial interstitium in the form of diffuse or nodular deposits. The most common findings are diffuse thickening of the interventricular septum (> 80% of patients) and the posterior wall of the LV. Isolated thickening of the wall of the right ventricle (without thickening of the LV wall) is rare - in 6% of cases, and thickening of the walls of both ventricles - in 40-80% of cases [3, 18]. Thickening of the interatrial septum is observed in 40% of patients. The change in the shape of the LV occurs according to the type of concentric hypertrophy; ventricular dilatation does not develop [19]. The combination of thickening of the myocardial walls in more than half of the patients with low amplitude of the ventricular complex on the electrocardiogram (ECG) in the chest leads (less than 10 mm) and limb leads (less than 5 mm) allows us to confirm the pseudohypertrophic nature of myocardial changes. According to C. Rapezzi et al., the thickness of the myocardial walls in patients with ATTR amyloidosis (especially senile) was significantly greater than in patients with AL amyloidosis [16, 20]. At the same time, patients with AL amyloidosis showed a more significant decrease in the amplitude of the ventricular complex and, accordingly, a lower ratio of QRS amplitude to LV wall thickness. This may indicate that the decrease in QRS amplitude in patients with AL amyloidosis is more related to damage to cardiomyocytes than to interstitial amyloid deposition. It is known that in the pathogenesis of organ damage in patients with AL amyloidosis, the direct toxic effect of free light chains of immunoglobulins on cells is of great importance [16, 18]. In patients with different types of AS, significant vacuolization of cardiomyocytes is detected. Studies in recent years have shown [24] that it is the vacuolization of cardiomyocytes, and not amyloid deposits, that is responsible for the increased echogenicity of the granular type, characteristic of AS and first described by A. Siqueira-Filho et al. [19]. Thus, this ultrasound property of amyloid heart is not specific and cannot be used as a diagnostic criterion.
Damage to cardiomyocytes in patients with AS may be reflected in increased concentrations of troponins and natriuretic peptides. The expression of atrial and medullary nitriuretic peptides has been shown to be increased in ventricular cardiomyocytes from patients with AS, particularly in areas adjacent to amyloid deposits [25]. Although these substances are not specific markers of amyloid cardiac damage, they are considered highly sensitive indicators and, therefore, an important prognostic factor in systemic amyloidosis [26].
Amyloid can be deposited in the area of the heart valves, often causing their thickening, noticeable on an echocardiogram [18, 27]. Thickening of the papillary muscles has been described [28], but valve function is often preserved.
Amyloid can damage the conduction system of the heart; cases of amyloid masses penetrating directly into the sinoatrial node have been described. Conduction disorders are most often represented by incomplete blockade of the anterior branch of the left bundle branch (20%), complete blockade of the right (4–19%) or left bundle branch (2–7%), and first degree atrioventricular block (18–33%) [16, 18]. Conduction disturbances, as a rule, occur in senile AS, which may be due to the longer course of this variant of the disease [3]. Amyloid deposition in the area of adrenergic synapses disrupts the neurohumoral regulation of heart function [29], which may be one of the risk factors for tachyarrhythmias. In 5–27% of patients, atrial fibrillation is detected; more rare rhythm disturbances are ventricular tachycardia and junctional rhythm. Some changes in the ECG occur in almost all (> 90%) patients with AS.
A distinctive property of AL amyloidosis is its deposition in the walls of the coronary arteries [15], which can cause myocardial ischemia [30]. Some patients complain of angina pectoris [31], although changes in the angiogram may be absent [32]. Myocardial infarction may develop. Pathological Q-waves and changes in repolarization without clinical signs of myocardial infarction or indication of it in the anamnesis are found in more than half of the patients, which allows these changes to be interpreted as pseudo-infarction. It appears that mimicking infarct changes is associated with nodular deposition of electrically inactive amyloid, which creates a scar effect. In the later stages of the disease, pericardial effusion is detected in almost half of patients [18].
Quantitative assessment of the total mass of amyloid deposits in the heart is possible using SAP scintigraphy. SAP is a normal plasma glycoprotein whose levels increase dramatically in amyloid deposits of any type as a result of reversible calcium-dependent binding to the amyloid fibril. After intravenous administration, the radiotracer labeled with SAP is distributed between the circulating and amyloid-bound SAP pools in proportion to their volume. In this way, images can be obtained for qualitative and quantitative assessment of amyloid deposits. The method is especially useful in monitoring the course of amyloidosis, including for the purpose of assessing the effectiveness of treatment.
Etiology of amyloidosis
The initiating factors for the development of primary amyloid renal dystrophy most often cannot be identified, and it itself often affects not only them, but also the tongue, dermis, thyroid gland, lungs, spleen, heart, liver and intestines.
There are often cases when the disease develops against the background of myeloma, characterized by a malignant course. Secondary renal amyloidosis affects not only the kidneys, but also the blood vessels, lymph nodes, liver and other internal organs. It develops in a number of pathological conditions:
- infectious nature (tuberculosis, malaria);
- purulent etiology (septic endocarditis, bronchiectasis);
- systemic (ankylosing spondylitis, rheumatoid arthritis);
- kidney (neoplastic);
- intestines (colitis, Crohn's disease).
The disease is detected in patients who have undergone extrarenal blood purification by hemodialysis for a long time.
Cardiac dysfunction
Intracardiac hemodynamic disturbances in AS are caused mainly by diastolic dysfunction; the decrease in myocardial contractility is usually insignificant. In a study that included 223 patients with AS, the mean LV ejection fraction was 52.5 ± 13.1% in patients with AL amyloidosis and 58 ± 13% in patients with hereditary ATTR amyloidosis. It was significantly lower only among patients with senile ATTR amyloidosis: 44.2 ± 15.4% [16]. A decrease in ejection fraction of less than 40% was noted in 40% of patients with senile ATTR amyloidosis and only in 22% of patients with AL amyloidosis and 8% of patients with hereditary ATTR amyloidosis. Other researchers have described similar changes in contractile function [18].
Diastolic dysfunction of varying degrees is detected in almost all patients. Early in the course of the disease, amyloid deposits impair isovolumic relaxation of the myocardium, resulting in a decrease in the rate of early diastolic LV filling (E) and an increase in late diastolic flow (A). Thus, a decrease in the E/A ratio (type 1 diastolic dysfunction) is an early sign of heart damage. As AS progresses, the myocardial wall becomes stiffer, the pressure in the left atrium increases, which leads to an increase in the rate of early diastolic filling and, thus, to a pseudo-normalization of the E/A ratio (type 2 diastolic dysfunction) [2]. With a further increase in the stiffness of the heart wall and an increase in end-diastolic pressure, a significant decrease in late diastolic flow occurs, which makes it possible to identify a decrease in myocardial compliance (3rd restrictive type of diastolic dysfunction). Restrictive changes are often accompanied by dilatation of both atria [3, 18].
Although restrictive disorders of diastolic function are considered the most typical for AS, research data show the possibility of developing other disorders of intracardiac hemodynamics [34–36].
Treatment
In the treatment of primary amyloidosis, glucocorticoid hormones and cytostatic drugs are used.
In secondary amyloidosis, treatment is aimed primarily at the underlying disease. Medicines of the 4-aminoquinoline series are also prescribed. A low-protein diet with limited salt is recommended.
The development of end-stage chronic renal failure is an indication for hemodialysis.
With the development of renal failure, hemodialysis is indicated
Clinical picture and prognosis
Amyloid damage to the heart in the early stages can be asymptomatic, manifested only by thickening of the LV wall on echocardiography [37]. Subsequently, clinical manifestations of HF with blood stagnation develop mainly in the systemic circulation [18]. Progressive shortness of breath and decreased exercise tolerance are observed in more than half of the patients and are accompanied by signs of pulmonary hypertension [2, 38]. In AL and senile ATTR amyloidosis, HF is more common (NYHA functional classes III–IV in more than half of the patients) than in hereditary ATTR amyloidosis [3, 16]. The systemic nature of the lesion is characteristic of AL amyloidosis. The majority develop damage to the kidneys (nephrotic syndrome, renal failure), liver (giant hepatomegaly, intrahepatic cholestasis), spleen (splenomegaly), muscles (macroglossia, muscle pseudohypertrophy), skin (periorbital purpura, amyloid plaques), peripheral and autonomic nervous system. Rarely, thromboembolism of the coronary arteries with amyloid masses develops, leading to death [39]. Thrombosis of various vessels has also been described [18].
The diagnosis of systemic amyloidosis is made by histological examination of tissue obtained from a biopsy. More often the wall of the rectum, stomach, oral cavity, subcutaneous fatty tissue, kidney and liver tissue are examined. Determination of the variant of amyloidosis is carried out using the immunohistochemical method; in case of hereditary amyloidosis, DNA analysis is done. If amyloidosis is limited to the heart, such as in hereditary ATTR amyloidosis with isoleucine mutation at position 122, then the only diagnostic method is myocardial biopsy. However, in most other cases, the diagnosis of AS can be established when amyloid is detected in a biopsy of another organ or tissue and the patient has a thickening of the LV wall of more than 12 mm according to echocardiography [40].
The most severe prognosis is for AL amyloidosis, and somewhat more favorable for ATTR amyloidosis. Moreover, the prognosis is determined precisely by heart damage. In a study of 232 patients with AL amyloidosis with cardiac involvement, their average life expectancy after diagnosis was 1 year. The presence of HF reduced survival: 0.75 versus 2.34 years in patients without HF.
Traditionally, cardiac predictors of poor prognosis, in addition to the severity of HF and restrictive disorders of LV diastolic function, are considered to be thickening and increase in LV mass with a decrease in ejection fraction [18, 41]. At the same time, C. Rapezzi et al. revealed the most significant thickening of the myocardial wall and increase in myocardial mass in patients with senile ATTR amyloidosis, in which relatively slow progression of AS is observed [16]. It is noteworthy that moderate thickening of the myocardial walls in patients with AL amyloidosis who participated in this study was combined with a significant decrease in the voltage of the ventricular ECG complexes. The authors associated these ECG changes with the direct toxic effect of immunoglobulin light chains on cardiomyocytes. At the same time, massive deposits of amyloid in patients with senile ATTR amyloidosis apparently do not have such an adverse effect on myocardial cells and, therefore, have less impact on the survival of patients. In another study of 57 patients with AL and ATTR amyloidosis, low ECG voltage was also associated with a worse prognosis [42].
Currently, high blood levels of markers of cardiomyocyte damage - troponins T and I, and N-terminal protein-brain natriuretic factor (NT-proBNP) at the time of diagnosis of AS are considered an important predictor of poor prognosis. Thus, in 261 patients with a newly diagnosed diagnosis, the average life expectancy with an increase in the level of troponins I or T was 6 and 8 months, respectively, and in the absence of troponins I or T in the blood - 21 and 22 months [43]. In this study, the prognostic value of serum troponin levels was higher than that of congestive heart failure and echocardiographic parameters. The possibility of analyzing the dynamics of NT-proBNP levels after treatment to assess the prognosis of AS is being studied. In AL amyloidosis, a 30% reduction in NT-proBNP levels after chemotherapy was associated with improved prognosis [44].
An important prognostic factor is also the presence and severity of extracardiac manifestations of systemic amyloidosis (orthostatic hypotension, complications of nephrotic syndrome and motor diarrhea, renal failure, etc.).
Thus, among the variants of AS, AL amyloidosis has the most severe and rapidly progressive course, in which the development of HF depends little on the degree of thickening of the myocardial walls and is not always combined with restrictive disorders of diastolic function. The progression of AS in these patients is largely due to damage to the cardiomyocyte. In patients with senile ATTR amyloidosis, a relatively mild course of cardiopathy is observed, despite significant structural (severe pseudohypertrophy) and functional (restrictive hemodynamic disorders) disorders. An intermediate position is occupied by hereditary ATTR amyloidosis, in which the symptoms of cardiopathy are close to those of AL amyloidosis of the heart, but are less malignant.
Forecast
Amyloidosis is a chronic, progressive disease. In secondary amyloidosis, the prognosis is largely determined by the possibility of treating the underlying disease.
As complications develop, the prognosis worsens. Once symptoms of heart failure appear, the average life expectancy is usually less than a few months. The life expectancy of patients with chronic renal failure is on average 12 months. This period increases slightly in the case of hemodialysis.
During pregnancy
Pregnancy with amyloidosis is allowed only if the functional activity of the woman’s vital organs allows her to bear and give birth to a healthy child. Otherwise, pregnancy can cause death not only for the fetus, but also for the mother herself.
Certain local forms of amyloidosis do not prevent the onset and gestation of pregnancy. Childbirth can proceed without complications and result in the birth of a healthy baby when amyloid protein accumulates in only one specific organ or tissue (intestinal wall, muscle tissue). In the generalized form, the prognosis for the mother and fetus is determined by the reserves of the affected organ and the duration of the disease.
Diagnosis of amyloid kidney dystrophy
During the latent stage, diagnosis of the disease is seriously difficult, so laboratory diagnostics of urine and blood are performed. The first make it possible to detect protein and leukocytes, the level of which is constantly increasing, as well as an admixture of blood and bodies from coagulated protein, blood cells, and kidney epithelium. Blood tests detect decreased albumin levels, increased globulin levels, abnormally elevated lipid and/or lipoprotein levels, and electrolyte imbalances.
In the presence of pathological processes, various diagnostic techniques make it possible to identify their specific manifestations. So:
- the electrocardiogram registers arrhythmia;
- echocardiography reveals myocardial pathologies, characterized by structural changes in the heart muscle;
- ultrasound diagnostics of the peritoneum determines an increase in the volume of internal organs such as the liver and spleen;
- X-ray examination of the gastrointestinal tract determines the deterioration of contraction of the stomach walls, too slow or, on the contrary, rapid movement of barium through the intestines;
- Ultrasound scanning of the kidneys reveals their significant increase;
A biopsy of the kidney, rectal mucosa and liver is required.
Complications
The severity of the disease depends on the location and extent of amyloid deposits. The most dangerous are neuropathic amyloidosis, amyloid damage to the kidneys and heart, against which the following complications often develop:
- renal failure;
- heart failure;
- paresis and paralysis (partial or complete limitation of mobility);
- dementia (dementia).
The disease can also lead to the formation of ulcers of the esophagus and stomach, gastrointestinal bleeding, liver failure, and diabetes mellitus.
Note! The likelihood of developing complications directly depends on the timeliness and adequacy of treatment. At the first signs of the disease, you should immediately consult a doctor and undergo a comprehensive examination.