Thalassemia (Cooley's anemia) is a hereditary disease in which the synthesis (production) of hemoglobin is impaired. Genetic pathology is more common among the peoples of Asia, the Near and Middle East, and African Americans; rare cases of the disease are registered in Russia in the Volga region. Clinical manifestations are varied and depend on the form of thalassemia. Sometimes the disease is diagnosed in the first days of life, in other cases it remains asymptomatic for a long time. Patients with mild forms live full lives and do not need drug therapy.
Classification
Thalassemia is classified according to the severity of the disease, mode of inheritance, and type of genetic defect. A detailed classification with explanations is presented in the table below.
| Classification sign | Form of the disease | Comments |
| Type of mutation | alpha thalassemia | associated with mutations in the HBA1 and HBA2 genes; with disturbances in one gene it is asymptomatic, changes in two or three genes cause chronic anemia, mutations in four genes are incompatible with life |
| beta thalassemia | distinguish beta thalassemia major and minor; the small form does not manifest itself in any way, the large form is characterized by a severe course | |
| Inheritance type | heterozygous | develops when inheriting one altered gene |
| homozygous | develops as a result of a mutation in both parents | |
| Severity of the disease | mild moderate severe | the severity of the disease depends on the number of mutated genes; in mild forms there are no symptoms, severe forms are often complicated by circulatory disorders and lead to the development of cardiovascular diseases |
Thalassemia is more common in regions where malaria is common. Researchers attribute this to the fact that carriers of this type of mutation in genes are more resistant to malarial plasmodium.
Genetic variants of alpha thalassemia
Based on the coverage of mutations in genes and their loci (certain parts, segments) responsible for the synthesis of alpha chains of polypeptides, it is proposed to distinguish several groups of the disease:
- mutation in only one locus - no clinical manifestations;
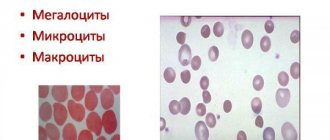
- changes in two (pairs) of loci in one or different genes - a blood test shows a decrease in hemoglobin, a decrease in the size of red blood cells;
- damage to three loci - is expressed in oxygen hypoxia of organs, enlargement of the spleen, the occurrence of H hemoglobinopathy, the resulting hemoglobin is unstable, decomposing, causing hemolytic anemia;
- mutation of all loci - completely stops the synthesis of alpha chains; in such a situation, intrauterine fetal death occurs or the child dies immediately after birth.
Genetic studies have also confirmed the unequal significance of pairs of genes, one pair of two is the main one, and the other has a secondary role. Clinical manifestations depend on which pair the mutation occurred in.
Bone marrow transplant is considered the most effective method
Causes of the disease
The cause of the development of the disease is a mutation in the gene, which carries information about the synthesis of globin chains - a protein involved in the formation of hemoglobin. As a result, its production (production) stops or occurs in smaller quantities, blood cells are destroyed, and anemia develops. The homozygous form of thalassemia is characterized by the presence of two defective genes that are inherited from both parents. With the heterozygous variant, a person becomes a carrier of an altered gene inherited from one of the parents.
Pathogenesis
The molecular basis of the pathogenesis of thalassemia is a genetic defect, which consists in the fact that in the cells of some patients abnormal RNA (ribonucleic acid, involved in coding, reading and regulation of genes) functions, while in others there is a deletion of genetic material (a change in the structure of chromosomes). As a result, the synthesis of one of the hemoglobin chains decreases or stops. Normally, the synthesis of hemoglobin chains is balanced, and the number of alpha and non-alpha chains is equal. A change in the synthesis of one of them leads to an imbalance. Thus, a lack of beta chains while maintaining the production of alpha chains leads to an excess of free alpha chains, and vice versa.
Genetic interpretation of beta thalassemia species
Genetic scientists have discovered an interesting pattern: people have the same mutation of the genes responsible for the synthesis of hemoglobin, but the clinical picture and severity of the disease differ.
Genes can be in the following states:
- normal - characteristic of a healthy person;
- partially damaged - “works” incompletely, which is why the synthesis of polypeptide chains is insufficient;
- completely destroyed - the synthesis stops.
On this basis, types of thalassemia are divided into:
- minor - the mildest form, only one gene is damaged, the person looks healthy outwardly, a blood test suggests slight anemia;
- intermedia - the lack of beta chains seriously affects the synthesis, red blood cells are underdeveloped, anemia is expressed with obvious signs, but the body can still adapt, so there is no need for constant blood transfusions;
- major - all genes have undergone mutations, the patient requires constant blood transfusions for health reasons.
Symptoms of Thalassemia
The clinical picture of thalassemia depends on the form of the disease. However, most patients have the following symptoms:
- delayed growth and development;
- change in skin color (pallor, yellowness, darkening);
- tendency to fractures due to thinning bones;
- skeletal deformities;
- enlarged liver and spleen;
- stones in the bile ducts.
In severe beta thalassemia, clinical manifestations are noticeable within the first year of a child’s life. Characteristic signs of this form of the disease:
- Mongoloid face (flattened bridge of the nose, narrowing of the palpebral fissures);
- altered head shape (“tower skull”);
- enlarged upper jaw;
- malocclusion.
Beta thalassemia minor is asymptomatic and is detected using laboratory diagnostic methods.
Note! Thalassemia can lead to mental and physical underdevelopment of the child, so the disease cannot be ignored. If you suspect a genetic pathology, you must undergo an examination and, if the diagnosis is confirmed, begin treatment.
Diagnosis of the disease
Pathology can be suspected in children with characteristic clinical signs in whose family cases of thalassemia have been identified. Patients with a suspected genetic mutation should visit a hematologist (blood disease specialist) and a medical geneticist. Laboratory diagnostics will help confirm or refute the diagnosis:
- Typical signs of thalassemia in a blood test are a decrease in hemoglobin levels and color index, the presence of red blood cells of an altered shape, an increase in iron and indirect bilirubin.
- When examining bone marrow substances, hyperplasia (proliferation) of the red hematopoietic lineage with a high number of erythroblasts and normoblasts (immature blood cells) is detected.
- Molecular genetic studies can identify a mutation in alpha or beta globin that disrupts the synthesis of globin protein chains.
- An ultrasound examination of the abdominal cavity reveals an enlarged liver and spleen, and stones in the gall bladder.
Differential diagnosis of the disease is carried out with iron deficiency, autoimmune, sickle cell and other types of anemia.
charitable foundation
The essence of the disease
Thalassemia is a group of hereditary diseases of the hematopoietic system, which are characterized by impaired hemoglobin synthesis. Therefore, the main manifestation of the disease is anemia.
As is known, normal human hemoglobin HbA consists of four protein chains of two different types: two α-chains and two β-chains. Accordingly, if the synthesis of α-chains is impaired (and abnormal hemoglobin consisting of four β-chains appears in the blood), then they speak of alpha thalassemia. If the synthesis of β-chains is impaired, then we are talking about beta-thalassemia; in this case, other variants of hemoglobin are formed that do not contain β-chains. Beta thalassemia is more common. There are also other variants of the disease, but their clinical significance is low.
The listed varieties of the disease are further divided into several subtypes depending on the number and type of genetic defects. Thus, beta thalassemia is divided into major, intermediate and minor forms; The most severe disease is thalassemia major, while beta thalassemia minor allows one to lead an almost normal life.
Frequency of occurrence, risk factors
Thalassemias are hereditary diseases. Severe forms of thalassemia are inherited by an autosomal recessive mechanism, that is, affected children can be born in those families where both the father and mother are carriers of the defective gene - however, the corresponding blood abnormalities in the parents are weakly expressed, since they also have “healthy” copies the required gene. Thus, two parents with beta thalassemia minor can have a child with thalassemia major.
The disease occurs in both boys and girls. Its frequency is about 1 in 100 thousand on average worldwide, but depends primarily on the region. Most patients with thalassemia are residents of southern countries (Mediterranean, Central and South Asian countries). This is due to the fact that carriage of a “defective” gene leads to better resistance to malaria. Therefore, in regions with widespread malaria, the number of carriers and, accordingly, the number of patients with thalassemia can be quite large. In Russia, there are relatively few patients with beta thalassemia major, and many of them are descendants of residents of the Volga region, the Caucasus or Central Asia.
For families who have already had children with severe forms of thalassemia, consultation with a geneticist is recommended. Since the carriage of the “defective” gene for beta thalassemia is easily determined by the results of simple and inexpensive blood tests, programs for the prevention of this disease are being developed in countries where thalassemia is widespread.
Signs and symptoms
With thalassemia, the usual symptoms of anemia are observed: pallor, shortness of breath, poor exercise tolerance, decreased appetite. In beta thalassemia major, anemia usually begins to appear at a few months of age.
Growth retardation is common in children. Abdominal pain may occur due to an enlarged spleen and gallstones. An enlarged liver, yellowness of the skin and mucous membranes, bone deformations (due to impaired functioning of the bone marrow), malocclusion, heart failure and/or arrhythmias, etc. are often observed.
Diagnostics
With thalassemia, characteristic changes are observed in the clinical blood test. The number of red blood cells, hemoglobin level (in severe forms - sometimes up to 20-30 g/l) and color index are sharply reduced. Red blood cells are reduced in size, often of an unusual appearance - the so-called “target-shaped”. For diagnosis, it is also useful to use the results of a biochemical blood test and, possibly, an analysis of a bone marrow sample.
Analysis of hemoglobin by electrophoresis, as well as biochemical determination of fetal hemoglobin, can confirm the diagnosis. In thalassemia, the amount of “normal” hemoglobin is reduced, but the amount of abnormal hemoglobin is increased.
It is useful to collect a family history to establish the hereditary nature of anemia. In selected cases, genetic testing may be performed to identify a specific genetic defect.
Treatment
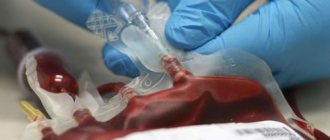
To treat anemia in thalassemia major, red blood cell transfusions are used - for example, once a month or two months. They allow you to maintain an acceptable hemoglobin level. However, regular transfusions lead to the accumulation of excess iron in the body - that is, iron overload. Gradually, this excess becomes life-threatening, and patients must take special chelating medications to remove iron from the body.
If the spleen is very enlarged (splenomegaly), surgical removal may be recommended. Enlargement of the spleen in thalassemia is associated both with its increased participation in the destruction of abnormal red blood cells, and with the processes of ineffective hematopoiesis occurring in it. Removing the spleen leads to some improvement, but increases the risk of infections and other complications. In recent years, information has emerged that some patients may benefit from therapy with the drug Jakavi (ruxolitinib): studies show that such therapy, if successful, leads to a decrease in the size of the spleen and a decrease in the need for transfusions, and also, in combination with immunosuppressive drugs, increases chances of success of subsequent transplantation.
Since we are talking about a genetically determined disease, no conservative therapy can lead to a complete cure. The only chance to normalize hematopoiesis is provided by allogeneic bone marrow transplantation. However, this is a complex procedure associated with a risk to life. If there is a compatible healthy donor among the patient's siblings, the transplant is usually successful. Transplantations from other donors are now being carried out more and more often, but they are associated with serious difficulties, primarily with an increased risk of rejection. Therefore, doctors are constantly developing new protocols for preparing for transplantation.
Forecast
Previously, severe forms of thalassemia (such as beta thalassemia major) usually resulted in death in early childhood. The situation has changed in recent decades. Now, periodic blood transfusions in combination with therapy aimed at removing excess iron often allow patients to live into middle age and even old age. However, it should be remembered that this therapy does not lead to a complete cure and must be lifelong.
Bone marrow transplantation, if successful, leads to normalization of hematopoiesis, but its implementation is associated with certain risks, especially if the patient does not have a compatible related donor.
Treatment of thalassemia
Treatment for thalassemia depends on the form of the disease. Patients with beta thalassemia minor do not require drug therapy other than taking B vitamins and folic acid. Patients with homozygous beta thalassemia from the first days of life are recommended to undergo blood transfusion therapy (transfusion of red blood cells), the introduction of chelating drugs (to bind iron), and hormones in case of a sharp deterioration of the condition. In severe cases, bone marrow transplantation is indicated for patients with thalassemia.
Epidemiology
Malaria
The worldwide distribution of hereditary alpha thalassemia is consistent with areas of malaria exposure suggesting a protective role. Thus, alpha thalassemia is common in sub-Saharan Africa. Africa, the Mediterranean basin, and tropical (and subtropical) regions in general. The epidemiology of alpha thalassemia in the United States reflects this global pattern of distribution. More specifically, HbH disease appears in Southeast Asia and the Middle East, while Hb Bart hydrops fetalis is recognized only in Southeast Asia.[21] Data shows that 15% of Greek and Turkish Cypriots are carriers of beta thalassemia. genes, and 10% of the population carry alpha thalassemia genes.[22]
Complications of the disease
In severe forms, the disease can lead to the development of complications. Among the most common complications of a genetic mutation of the globin protein are:
- Increased iron levels in the blood. Excess iron in the body occurs due to the progression of the disease or as a result of frequent blood transfusions. High levels of iron lead to damage to the liver, endocrine system, and cause the development of arrhythmia.
- Enlarged spleen (splenomegaly). This genetic mutation is often accompanied by the destruction of a large number of red blood cells, creating an increased load on the spleen, as a result of which the organ increases in size. Splenomegaly can worsen anemia and reduce life expectancy. If the spleen is very enlarged, surgery to remove it - splenectomy - may be required. Surgery is recommended for patients over six years of age.
- Infectious complications. With a genetic mutation, the likelihood of developing infectious diseases increases. The highest risk is observed in patients after removal of the spleen.
- Bone deformation. Thalassemia can lead to bone marrow overgrowth and bone deformities. Enlarged bone marrow also causes thinning bones and increases the likelihood of fractures.
Etiology
Thalassemia is caused by point mutations or deletions in the genes encoding hemoglobin chains. As a result, this can lead to a decrease in synthesis or complete absence of one of the chains in the body. The other chain forms inadequate hemoglobin tetramers, which leads to the destruction of red blood cells and hemolytic anemia.
The structure of hemoglobin
Hemoglobin is a protein found in red blood cells that is responsible for transporting oxygen to tissues and carbon dioxide from them.
Hemoglobin (we are talking about HbA) consists of four chains: two alpha subunits and two beta subunits. This hemoglobin makes up 97% of the total content in red blood cells.
The remaining 3% is hemoglobin HbA2, which differs in the structure of its two chains: instead of beta subunits, it has delta subunits. HbA and HbA2 are normal when they are in the correct ratio.
Each of the hemoglobin chains binds to its non-protein part - heme.
So, with thalassemia, the synthesis of one of the hemoglobin chains is disrupted: either alpha or beta. According to this principle, there is a classification of thalassemia into:
- alpha thalassemia;
- beta thalassemia.
Thalassemia is classified according to severity:
- mild;
- moderate;
- severe.
Types of hemoglobin
There are physiological and pathological types of hemoglobin.
Hemoglobin, which may be normal in humans, includes:
- HbP – primitive hemoglobin, found in the embryo between the 7th and 12th weeks of life;
- HbF – fetal hemoglobin, contains two alpha and two gamma chains, appears after 12 weeks of intrauterine development, in adults its content is less than 1%;
- HbA – hemoglobin of adults, the proportion is 97%, contains two alpha and two beta chains;
- HbA2 – hemoglobin of adults, the proportion is 2%, contains two alpha and two delta chains,
- HbO2 – oxyhemoglobin, formed when oxygen binds in the lungs;
- HbCO2 – carbohemoglobin, is formed when carbon dioxide binds in tissues.
Pathological forms of hemoglobin include:
- HbS – hemoglobin, determined in sickle cell anemia;
- MetHb – methemoglobin, contains a trivalent iron ion, when normally it is divalent. This form is formed when consuming sulfonamides, nitrates, or vitamin C deficiency. Methemoglobin is not able to bind oxygen, resulting in tissue hypoxia;
- HbCO – carboxyhemoglobin, is formed when there is excess carbon monoxide in the inhaled air. It is present in small concentrations in the blood, but its level may increase depending on the characteristics of the inhaled air.
HbS is a type of hemoglobin that occurs when the beta chain is mutated (one amino acid is replaced by another). It occurs in people with sickle cell anemia. Red blood cells containing such hemoglobin do not live long and are quickly destroyed, which is good in the habitats of the malarial plasmodium. People with sickle cell disease have resistance to this parasite.
Prevention of thalassemic mutations
To prevent the risk of having children with thalassemia, parents need to visit a medical geneticist at the stage of pregnancy planning. This is especially true for families with genetic mutations of varying severity in their family. Modern antenatal diagnostics using high-tech equipment makes it possible to identify most thalassemic mutations.
If thalassemia is detected after the birth of the child, it is important to begin treatment on time. Timely adequate therapy will reduce the risk of complications and bone deformities in a child to a minimum.
Recommendations
- ^ a b c d f g gram hour
Origa, Rafaella;
Mine, Paolo; Galanello, Renzo; Cao, Antonio (1 January 1993). "Alpha thalassemia." GeneReviews
. PMID 20301608. Received September 22, 2021. update 2013 - ^ a b BRS Pathology
(4th ed.). Lippincott Williams and Wilkins Medical. December 2009 p. 162. ISBN 978-1451115871. - ^ a b c d f
"Examination for alpha thalassemia: approaches, laboratory tests, hemoglobin electrophoresis."
emedicine.medscape.com
. Retrieved May 24, 2021. - ^ a b
“Complications and treatment |
Thalassemia | Blood diseases | NCBDDD | CDC." www.cdc.gov
. Retrieved September 22, 2021. - Online Mendelian Inheritance in Man (OMIM): Hemoglobin - Alpha locus 1; HBA1 - 141800
- Online Mendelian Inheritance in Man (OMIM): Hemoglobin - Alpha Locus 2; HBA2 - 141850
- Langkowski's Guide to Pediatric Hematology and Oncology, 6th edition (2016)
. - ^ a b c d f
“Alpha thalassemia - Symptoms, diagnosis and treatment |
BMJ Best Practice". bestpractice.bmj.com
. Retrieved November 17, 2021. - Help, House of Genetics. "Alpha thalassemia." A Home Guide to Genetics
. Retrieved November 25, 2021. - Origa, Rafaella; Moi, Paolo (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (ed.), "Alpha Thalassemia", GeneReviews®
, University of Washington, Seattle, PMID 20301608, retrieved November 25, 2019 - "Assessment of Anemia - Etiology | BMJ Best Practices.” bestpractice.bmj.com
. Retrieved November 25, 2021. - Steensma D.P., Gibbons R.J., Higgs D.R. (January 2005). "Acquired alpha thalassemia in association with myelodysplastic syndrome and other hematologic malignancies." Blood
.
105
(2):443–52. doi:10.1182/blood-2004-07-2792. PMID 15358626. - ^ a b
Galanello R., Cao A. (February 2011).
“Gene test review. Alpha thalassemia." Genetics in Medicine
.
13
(2): 83–8. Doi:10.1097/GIM.0b013e3181fcb468. PMID 21381239. - "Hemoglobin H disease." Orphanet
. Retrieved September 22, 2021. - Vichinsky E.P. (January 1, 2009). "Alpha thalassemia major—new mutations, in utero management and outcomes." Hematology.
American Society of Hematology. Educational program .
2009
(1): 35–41. doi:10.1182/asheducation-2009.1.35. PMID 20008180. - ^ a b c
Viprakasit, VIP;
Equattanakit, Supachai (April 1, 2021). "Clinical classification, screening and diagnosis of thalassemia." Hematology/Oncology Clinics of North America
.
Thalassemia. 32
(2): 193–211. doi:10.1016/j.hoc.2017.11.006. ISSN 0889-8588. PMID 29458726. - "In a timely manner". www.uptodate.com
. Retrieved November 25, 2021. - ^ a b
Taher, Ali;
Musallam, Khaled; Cappellini, Maria Domenica, ed. (2017). Guidelines for the management of non-transfusion-dependent thalassemia (NTDT)
(2nd ed.). International Thalassemia Foundation. pp. 24–32. Retrieved November 5, 2021. - "Thalassemia | Doctor | Patient". Patient
. Retrieved September 22, 2021. - Kreger EM, Singer ST, Witt RG, Sweeters N, Lianoglou B, Lal A, et al (December 2021). "Favorable outcomes after intrauterine transfusion in fetuses with alpha thalassemia major: a case series and review of the literature." Prenatal diagnosis
.
36
(13):1242–1249. Doi:10.1002/pd.4966. PMID 27862048. - Harteveld K.L., Higgs D.R. (May 2010). "Alpha thalassemia." Orphanet Journal of Rare Diseases
.
5
(1): 13. doi:10.1186/1750-1172-5-13. PMC 2887799. PMID 20507641. - Hematology made simpler. AuthorHouse. 2013-02-06. ISBN 9781477246511.p. 246