Талассемия (анемия Кули) – наследственное заболевание, при котором нарушается синтез (выработка) гемоглобина. Генетическая патология чаще встречается у народов Азии, Ближнего и Среднего Востока, афроамериканцев, редкие случаи болезни регистрируются в России на территории Поволжья. Клинические проявления разнообразны и зависят от формы талассемии. Иногда заболевание диагностируется в первые дни жизни, в других случаях долго протекает бессимптомно. Больные с легкими формами живут полной жизнью и не нуждаются в лекарственной терапии.
Классификация
Талассемию классифицируют по тяжести заболевания, типу наследования и виду генетического дефекта. Подробная классификация с пояснениями представлена ниже в таблице.
| Признак классификации | Форма заболевания | Комментарии |
| Вид мутации | альфа-талассемия | связана с мутациями в генах HBA1 и HBA2; при нарушениях в одном гене протекает бессимптомно, изменения двух или трех генов вызывают хроническую анемию, мутации четырех генов несовместимы с жизнью |
| бета-талассемия | выделяют большую и малую бета-талассемии; малая форма никак не проявляется, большая форма характеризуется тяжелым течением | |
| Тип наследования | гетерозиготная | развивается при наследовании одного измененного гена |
| гомозиготная | развивается при мутации у обоих родителей | |
| Тяжесть заболевания | легкая средней степени тяжелая | степень тяжести заболевания зависит от количества мутировавших генов; при легких формах симптомы отсутствуют, тяжелые формы часто осложняются нарушениями кровообращения и приводят к развитию сердечно-сосудистых заболеваний |
Талассемия чаще встречается в регионах, где распространена малярия. Исследователи связывают это с тем, что носители данного вида мутаций в генах более устойчивы к малярийному плазмодию.
Генетические разновидности альфа-талассемии
По охвату мутацией генов, их локусов (определенных частей, отрезков), отвечающих за синтез альфа-цепочек полипептидов, предлагается выделение нескольких групп заболевания:
- мутация только в одном локусе — клинических проявлений нет;
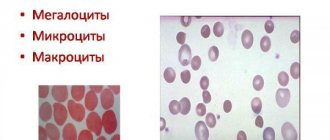
- изменения двух (пары) локусов в одном или разных генах — анализ крови показывает снижение гемоглобина, уменьшение размера эритроцитов;
- поражение трех локусов — выражается в кислородной гипоксии органов, увеличении селезенки, возникновении Н гемоглобинопатии, образующийся гемоглобин нестоек, разлагаясь, вызывает гемолитическую анемию;
- мутация всех локусов — полностью прекращает синтез альфа-цепей, при подобной ситуации происходит внутриутробная гибель плода или ребенок умирает сразу после родов.
Генетические исследования подтвердили также неодинаковое значение пар генов, одна пара из двух является главной, а другая имеет второстепенную роль. Клинические проявления зависят от того, в какой паре произошла мутация.
Пересадка костного мозга считается наиболее результативным способом
Причины заболевания
Причина развития болезни – мутация в гене, который несет информацию о синтезе цепей глобина – белка, участвующего в образовании гемоглобина. Вследствие чего его продуцирование (выработка) прекращается либо происходит в меньшем количестве, клетки крови разрушаются, и развивается анемия. При гомозиготной форме талассемии характерно наличие двух дефектных генов, которые наследуются от обоих родителей. При гетерозиготном варианте человек становится носителем измененного гена, унаследованного от одного из родителей.
Патогенез
Молекулярную основу патогенеза талассемии составляет генетический дефект, который заключается в том, что в клетках у одних пациентов функционирует аномальная РНК (рибонуклеиновая кислота, участвует в кодировании, прочтении и регуляции генов), а у других происходит делеция генетического материала (изменение в структуре хромосом). Вследствие этого снижается или прекращается синтез одной из цепочек гемоглобина. В норме синтез цепей гемоглобина сбалансирован, а количество альфа- и не альфа-цепей одинаково. Изменение синтеза одной из них приводит к нарушению баланса. Так, недостаток бета-цепей при сохранении продукции альфа-цепей приводит к избытку свободных альфа-цепей, и наоборот.
Генетическая интерпретация видов бета-талассемии
Ученые-генетики выяснили интересную закономерность: люди имеют одинаковую мутацию генов, отвечающих за синтез гемоглобина, но клиника и степень выраженности заболевания у них отличаются.
Гены могут находиться в следующих состояниях:
- нормальные — характерны для здорового человека;
- поврежденные частично — «работает» неполно, из-за чего синтез полипептидных цепей недостаточен;
- разрушенные полностью — синтез останавливается.
По этому признаку виды талассемии делят на:
- минор — самая легкая форма, поврежден только один ген, внешне человек выглядит здоровым, по анализу крови можно предположить небольшую анемию;
- интермедиа — недостаток бета-цепей серьезно сказывается на синтезе, эритроциты недоразвиты, малокровие выражено с явными признаками, но организм еще может приспособиться, поэтому отсутствует необходимость в постоянных переливаниях крови;
- майор — мутации подверглись все гены, больному необходимы постоянные переливания крови по жизненным показаниям.
Симптомы талассемии
Клиническая картина талассемии зависит от формы заболевания. Однако для большинства пациентов характерны следующие признаки:
- задержка роста и развития;
- изменение цвета кожи (бледность, желтушность, потемнение);
- склонность к переломам из-за истончения костей;
- деформации скелета;
- увеличение печени и селезенки;
- камни в желчных путях.
При тяжелой форме бета-талассемии клинические проявления заметны уже в течение первого года жизни ребенка. Характерные признаки этой формы заболевания:
- монголоидное лицо (уплощенная переносица, сужение глазных щелей);
- измененная форма головы («башенный череп»);
- увеличенная верхняя челюсть;
- неправильный прикус.
Малая форма бета-талассемии протекает бессимптомно, и выявляется при помощи методов лабораторной диагностики.
Обратите внимание! Талассемия может привести к умственному и физическому недоразвитию ребенка, поэтому заболевание нельзя игнорировать. При подозрении на генетическую патологию нужно обязательно пройти обследование, и при подтверждении диагноза начать лечение.
Диагностика заболевания
Патологию можно заподозрить у детей с характерными клиническими признаками, в роду которых были выявлены случаи талассемии. Пациентам с подозрением на генетическую мутацию необходимо посетить врача-гематолога (специалиста по болезням крови) и медицинского генетика. Подтвердить или опровергнуть диагноз поможет лабораторная диагностика:
- Типичными признаками талассемии в анализе крови служат снижение уровня гемоглобина и цветового показателя, наличие эритроцитов измененной формы, повышение показателей железа и непрямого билирубина.
- При исследовании вещества костного мозга обнаруживается гиперплазия (разрастание) красного кроветворного ростка с высоким числом эритробластов и нормобластов (незрелых клеток крови).
- Молекулярно-генетические исследования позволяют выявить мутацию альфа- или бета-глобина, нарушающую синтез цепочек белка глобина.
- При ультразвуковом исследовании (УЗИ) брюшной полости выявляется увеличение печени и селезенки, обнаруживаются камни в желчном пузыре.
Дифференциальная диагностика заболевания проводится с железодефицитной, аутоиммунной, серповидно-клеточной и другими видами анемий.
благотворительный фонд
Суть болезни
Талассемии – группа наследственных заболеваний кроветворной системы, которые характеризуются нарушением синтеза гемоглобина. Поэтому основным проявлением болезни является анемия.
Как известно, нормальный человеческий гемоглобин HbA состоит из четырех белковых цепей двух разных видов: две α-цепи и две β-цепи. Соответственно, если нарушен синтез α-цепей (при этом в крови появляется аномальный гемоглобин, состоящий из четырех β-цепей), то говорят об альфа-талассемии. Если же нарушен синтез β-цепей, то речь идет о бета-талассемии; при этом образуются другие варианты гемоглобина, которые не содержат β-цепей. Чаще встречается именно бета-талассемия. Существуют также другие варианты болезни, но их клиническая значимость невелика.
Перечисленные разновидности болезни подразделяются еще на несколько подтипов в зависимости от числа и вида генетических дефектов. Так, у бета-талассемии выделяют большую, промежуточную и малую формы; наиболее тяжелым заболеванием является большая талассемия, тогда как малая форма бета-талассемии позволяет вести практически нормальную жизнь.
Частота встречаемости, факторы риска
Талассемии – наследственные заболевания. Тяжелые формы талассемии наследуются по аутосомно-рецессивному механизму, то есть больные дети могут родиться в тех семьях, где и отец, и мать являются носителями дефектного гена – однако соответствующие аномалии крови у родителей выражены слабо, так как у них есть и «здоровые» копии необходимого гена. Так, у двух родителей с малой формой бета-талассемии может родиться ребенок с большой талассемией.
Болезнь встречается как у мальчиков, так и у девочек. Частота ее составляет около 1 на 100 тысяч в среднем по всему миру, но зависит прежде всего от региона. Большинство больных талассемией – жители южных стран (Средиземноморье, страны Центральной и Южной Азии). Это связано с тем, что носительство «дефектного» гена ведет к лучшей сопротивляемости заболеванию малярией. Поэтому в регионах с широким распространением малярии число носителей и, соответственно, число больных талассемией может быть довольно велико. В России больных большой бета-талассемией сравнительно мало, при этом многие из них – потомки жителей Поволжья, Кавказа или Средней Азии.
Семьям, где уже были случаи рождения детей с тяжелыми формами талассемии, рекомендуется консультация генетика. Так как носительство «дефектного» гена при бета-талассемии легко определяется по результатам простых и недорогих анализов крови, в странах с широким распространением талассемии развиваются программы профилактики этого заболевания.
Признаки и симптомы
При талассемии наблюдаются обычные для анемии симптомы: бледность, одышка, плохая переносимость физических нагрузок, сниженный аппетит. При большой форме бета-талассемии анемия обычно начинает проявляться в возрасте нескольких месяцев.
У детей нередко встречается задержка роста. Могут возникать боли в животе из-за увеличения селезенки и камней в желчном пузыре. Нередко наблюдаются увеличение печени, желтушность кожи и слизистых оболочек, деформации костей (из-за нарушения функционирования костного мозга), аномалии прикуса, сердечная недостаточность и/или аритмии и др.
Диагностика
При талассемии наблюдаются характерные изменения в клиническом анализе крови. Резко снижены количество эритроцитов, уровень гемоглобина (при тяжелых формах – иногда до 20–30 г/л) и цветовой показатель. Эритроциты крови уменьшенного размера, зачастую необычного вида – так называемые «мишеневидные». Для диагностики полезно использовать также результаты биохимического исследования крови и, возможно, анализ образца костного мозга.
Анализ гемоглобина методом электрофореза, а также биохимическое определение фетального гемоглобина позволяют подтвердить диагноз. При талассемиях снижено количество «нормального» гемоглобина, но повышено количество аномального.
Полезен сбор семейного анамнеза для установления наследственной природы анемии. В отдельных случаях может проводиться генетический анализ для выявления конкретного генетического дефекта.
Лечение
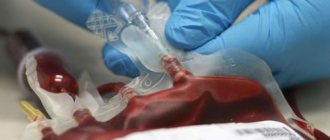
Для лечения анемии при большой форме талассемии применяются переливания эритроцитарной массы – например, раз в месяц или два месяца. Они позволяют поддерживать приемлемый уровень гемоглобина. Однако регулярные переливания приводят к накоплению в организме избытка железа – то есть перегрузке железом. Постепенно этот избыток становится опасным для жизни, и больные должны принимать специальные лекарства-хелаторы для выведения железа из организма.
Если сильно увеличена селезенка (спленомегалия), может быть рекомендовано ее хирургическое удаление. Увеличение селезенки при талассемии связано как с ее повышенным участием в разрушении аномальных эритроцитов, так и с происходящими в ней процессами неэффективного кроветворения. Удаление селезенки приводит к некоторому улучшению состояния, но повышает риск инфекций и других осложнений. В последние годы появилась информация, что части пациентов может помочь терапия препаратом «Джакави» (руксолитиниб): исследования показывают, что такая терапия в случае успеха приводит к уменьшению размеров селезенки и снижению потребности в переливаниях, а также, в сочетании с иммуносупрессивными препаратами, повышает шансы на успех последующей трансплантации.
Так как речь идет о генетически обусловленном заболевании, никакая консервативная терапия не может привести к полному излечению. Единственный шанс на нормализацию кроветворения дает аллогенная трансплантация костного мозга. Однако это сложная процедура, связанная с риском для жизни. При наличии совместимого здорового донора среди братьев и сестер пациента трансплантация обычно проходит успешно. Трансплантации от других доноров сейчас проводятся все чаще, но они бывают сопряжены с серьезными трудностями, прежде всего с повышенным риском отторжения. Поэтому врачи все время разрабатывают новые протоколы подготовки к трансплантации.
Прогноз
Раньше тяжелые формы талассемии (такие как большая форма бета-талассемии) обычно приводили к смерти в раннем детском возрасте. Ситуация изменилась в последние десятилетия. Сейчас периодические переливания крови в сочетании с терапией, направленной на удаление избытка железа, часто позволяют больным доживать до среднего и даже пожилого возраста. Однако следует помнить, что эта терапия не ведет к полному излечению и должна быть пожизненной.
Трансплантация костного мозга в случае успеха приводит к нормализации кроветворения, но ее проведение связано с определенными рисками, особенно есть у больного нет совместимого родственного донора.
Лечение талассемии
Лечение талассемии зависит от формы заболевания. Больные с малой бета-талассемией не нуждаются в лекарственной терапии, кроме приема витаминов группы B и фолиевой кислоты. Пациентам с гомозиготной бета-талассемией с первых дней жизни показано проведение гемотрансфузионной терапии (переливание эритроцитарной массы), введение хелатирующих препаратов (для связывания железа), гормонов при резком ухудшении состояния. В тяжелых случаях больным с талассемией показана трансплантация костного мозга.
Эпидемиология
Малярия
Распространение наследственной альфа-талассемии во всем мире соответствует областям малярия воздействие, предполагающее защитную роль. Таким образом, альфа-талассемия распространена в странах к югу от Сахары. Африка, то Средиземноморский бассейн, и вообще тропические (и субтропические) регионы. Эпидемиология альфа-талассемии в США отражает эту глобальную модель распространения. Более конкретно, болезнь HbH проявляется в Юго-Восточная Азия и Средний Восток, в то время как Hb Bart hydrops fetalis признан только в Юго-Восточной Азии.[21]Данные показывают, что 15% Греческий и Турки-киприоты являются носителями бета-талассемия гены, а 10% населения несут гены альфа-талассемии.[22]
Осложнения заболевания
При тяжелых формах заболевание может приводить к развитию осложнений. Среди наиболее частых осложнений генетической мутации белка глобина встречаются:
- Повышение уровня железа в крови. Избыток железа в организме возникает вследствие прогрессирования заболевания либо в результате частых переливаний крови. Высокий уровень железа приводит к повреждению печени, эндокринной системы, становится причиной развития аритмии.
- Увеличение селезенки (спленомегалия). Данная генетическая мутация часто сопровождается разрушением большого числа эритроцитов, создавая повышенную нагрузку на селезенку, вследствие чего орган увеличивается в размерах. Спленомегалия может усугубить течение анемии и снизить продолжительность жизни. При большом увеличении селезенки может потребоваться хирургическая операция по её удалению – спленэктомия. Оперативное вмешательство рекомендуется проводить пациентам старше шести лет.
- Инфекционные осложнения. При генетической мутации повышается вероятность развития инфекционных заболеваний. Наиболее высокий риск отмечается у пациентов после удаления селезенки.
- Деформация костей. Талассемия может привести к разрастанию костного мозга и деформации костей. Увеличение костного мозга также способствует истончению костей и повышению вероятности переломов.
Этиология
Причиной талассемии являются точечные мутации или делеции в генах, кодирующих цепи гемоглобина. В результате это может привести к уменьшению синтеза или полному отсутствию одной из цепей в организме. Другая цепь образует неадекватные тетрамеры гемоглобина, что приводит к разрушению эритроцитов и гемолитической анемии.
Строение гемоглобина
Гемоглобин – белок содержащийся в эритроцитах, отвечающий за перенос кислорода к тканям и углекислого газа от них.
Гемоглобин (речь идёт о HbA) состоит из четырёх цепей: двух альфа-субъединиц и двух бета-субъединиц. Такой гемоглобин составляет 97% от общего содержания его в эритроцитах.
Оставшиеся 3% составляет гемоглобин HbA2, отличающийся по строению двух цепей: вместо бета-субъединиц у него дельта-субъединицы. HbA и HbA2 является вариантом нормы при их правильном соотношении.
Каждая из цепей гемоглобина связывается с его небелковой частью – гемом.
Так вот при талассемии нарушается синтез одной из цепей гемоглобина: либо альфа, либо бета. По этому принципу имеется классификация талассемии на:
- альфа-талассемию;
- бета-талассемию.
По степени тяжести выделяют талассемию:
- лёгкой степени;
- средней степени;
- тяжёлой степени.
Разновидности гемоглобина
Существуют физиологические виды гемоглобина и патологические.
К гемоглобину, который может быть в норме у человека, относится:
- HbP – примитивный гемоглобин, встречается в эмбрионе между 7 – 12-й неделей жизни;
- HbF – фетальный гемоглобин, содержит две альфа и две гамма-цепи, появляется через 12 недель внутриутробного развития, у взрослых его содержание составляет менее 1%;
- HbA – гемоглобин взрослых, доля составляет 97%, содержит две альфа и две бета-цепи;
- HbA2 – гемоглобин взрослых, доля составляет 2%, содержит две альфа и две дельта-цепи,
- HbO2 – оксигемоглобин, образуется при связывании кислорода в лёгких;
- HbCO2 – карбогемоглобин, образуется при связывании углекислого газа в тканях.
К патологическим формам гемоглобина относятся:
- HbS – гемоглобин, определяемый при серповидноклеточной анемии;
- MetHb – метгемоглобин, содержит трёхвалентный ион железа, когда в норме оно двухвалентное. Такая форма образуется при употреблении сульфаниламидов, нитратов, дефиците витамина С. Метгемоглобин не способен связывать кислород, в результате чего возникает гипоксия тканей;
- HbCO – карбоксигемоглобин, образуется при наличии избытка угарного газа во вдыхаемом воздухе. В крови он присутствует в небольших концентрациях, но его уровень может повышаться в зависимости от характеристики вдыхаемого воздуха.
HbS – вид гемоглобина, возникающего при мутации бета-цепи (одна аминокислота заменяется на другую). Он образуется у людей с серповидноклеточной анемией. Эритроциты, содержащие такой гемоглобин, долго не живут и быстро разрушаются, что хорошо в местах обитания малярийного плазмодия. У лиц с серповидноклеточной анемией имеется устойчивость к этому паразиту.
Профилактика талассемических мутаций
Для предупреждения риска рождения детей с талассемией родителям необходимо еще на этапе планирования беременности посетить медицинского генетика. Особенно это актуально для семей, имеющих в роду генетические мутации той или иной степени выраженности. Современная антенатальная диагностика с использованием высокотехнологичного оборудования позволяет выявить большинство талассемических мутаций.
Если талассемия выявлена уже после рождения ребенка, важно вовремя начать ее лечение. Своевременная адекватная терапия сведет риск развития осложнений и костных деформаций у ребенка к минимуму.
Рекомендации
- ^ абcdежграммчас
Орига, Рафаэлла; Мои, Паоло; Галанелло, Ренцо; Цао, Антонио (1 января 1993 г.). «Альфа-талассемия».
GeneReviews
. PMID 20301608. Получено 22 сентября 2021.обновление 2013 - ^ абBRS Патология
(4-е изд.). Липпинкотт Уильямс и Уилкинс медицинский. Декабрь 2009 г. с. 162. ISBN 978-1451115871 . - ^ абcdе
«Обследование при альфа-талассемии: подходы, лабораторные исследования, электрофорез гемоглобина».
emedicine.medscape.com
. Получено 24 мая 2021. - ^ аб
«Осложнения и лечение | Талассемия | Заболевания крови | NCBDDD | CDC».
www.cdc.gov
. Получено 22 сентября 2021. - Онлайн-менделевское наследование в человеке (OMIM): Гемоглобин — Альфа-локус 1; HBA1 — 141800
- Онлайн-менделевское наследование в человеке (OMIM): Гемоглобин — Альфа-локус 2; HBA2 — 141850
- Руководство Ланжковского по детской гематологии и онкологии, 6-е издание (2016 г.)
. - ^ абcdе
«Альфа-талассемия — Симптомы, диагностика и лечение | BMJ Best Practice».
bestpractice.bmj.com
. Получено 17 ноября 2021. - Справка, Дом генетики. «Альфа-талассемия». Домашний справочник по генетике
. Получено 25 ноября 2021. - Орига, Рафаэлла; Мои, Паоло (1993), Адам, Маргарет П .; Ardinger, Holly H .; Пагон, Роберта А .; Уоллес, Стефани Э. (ред.), «Альфа-талассемия», GeneReviews®
, Вашингтонский университет, Сиэтл, PMID 20301608, получено 25 ноября 2019 - «Оценка анемии — Этиология | Лучшие практики BMJ». bestpractice.bmj.com
. Получено 25 ноября 2021. - Стинсма Д.П., Гиббонс Р.Дж., Хиггс Д.Р. (январь 2005 г.). «Приобретенная альфа-талассемия в сочетании с миелодиспластическим синдромом и другими гематологическими злокачественными новообразованиями». Кровь
.
105
(2): 443–52. Дои:10.1182 / кровь-2004-07-2792. PMID 15358626. - ^ аб
Галанелло Р., Цао А. (февраль 2011 г.). «Обзор генного теста. Альфа-талассемия».
Генетика в медицине
.
13
(2): 83–8. Дои:10.1097 / GIM.0b013e3181fcb468. PMID 21381239. - «Болезнь гемоглобина H». Orphanet
. Получено 22 сентября 2021. - Вичинский Е.П. (1 января 2009 г.). «Большая альфа-талассемия — новые мутации, внутриутробное ведение и исходы». Гематология. Американское общество гематологии. Образовательная программа
.
2009
(1): 35–41. Дои:10.1182 / asheducation-2009.1.35. PMID 20008180. - ^ абc
Випракасит, VIP; Экваттанакит, Супачай (1 апреля 2021 г.). «Клиническая классификация, скрининг и диагностика талассемии».
Гематологические / онкологические клиники Северной Америки
. Талассемия.
32
(2): 193–211. Дои:10.1016 / j.hoc.2017.11.006. ISSN 0889-8588. PMID 29458726. - «Своевременно». www.uptodate.com
. Получено 25 ноября 2021. - ^ аб
Тахер, Али; Мусаллам, Халед; Каппеллини, Мария Доменика, ред. (2017).
Рекомендации по ведению нетрансфузионно-зависимой талассемии (NTDT)
(2-е изд.). Международный фонд талассемии. стр. 24–32. Получено 5 ноября 2021. - «Талассемия | Врач | Пациент». Пациент
. Получено 22 сентября 2021. - Kreger EM, Singer ST, Witt RG, Sweeters N, Lianoglou B, Lal A и др. (Декабрь 2021 г.). «Благоприятные исходы после внутриутробного переливания у плодов с большой альфа-талассемией: серия случаев и обзор литературы». Пренатальная диагностика
.
36
(13): 1242–1249. Дои:10.1002 / pd.4966. PMID 27862048. - Хартевельд К.Л., Хиггс Д.Р. (май 2010 г.). «Альфа-талассемия». Журнал редких заболеваний Orphanet
.
5
(1): 13. Дои:10.1186/1750-1172-5-13. ЧВК 2887799. PMID 20507641. - Гематология стала проще. АвторДом. 2013-02-06. ISBN 9781477246511.стр. 246