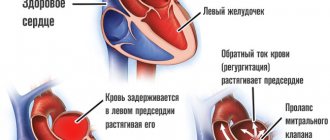
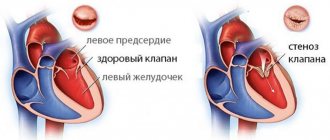
Физиологическая гипертрофия[править | править код]
Для физиологической гипертрофии сердца у спортсменов типичны следующие признаки: 1) определенные амплитудные характеристики зубцов комплекса QRS; 2) отсутствие расширения QRS; 3) нормальное время местной электронегативности; 4) отсутствие нарушений деполяризации смешений книзу сегмента S-Т, деформаций или инверсии зубца Т; 5) увеличение объема сердца свыше 11 мл/кг.
На ЭКГ фиксируются признаки гипертрофии преимущественно левого желудочка. Наиболее чувствительными электрокардиографическими показателями физиологической гипертрофии левого желудочка у спортсменов в порядке убывающей точности являются: Rv5-6 > 33 мм, SV1 + Rv5-6> 53 мм, сумма амплитуд QRS в отведениях V2, V5, aVF > 93 мм, Ravf 23 мм, Rmax + Smax в грудных отведениях > 55 мм.
Наиболее чувствительными электрокардиографическими показателями физиологической гипертрофии правого желудочка у спортсменов в порядке убывающей точности являются: Rv1 + Sv5 > 11 мм, SV5-6 > 7 мм, Rv1 > 7 мм. Электрокардиографические признаки гипертрофии правого желудочка менее надежны, чем признаки гипертрофии левого желудочка. Сочетание на ЭКГ нескольких признаков гипертрофии позволяет с большей уверенностью говорить о ее наличии.
Возможно использование отдельных индексов. Так, при гипертрофии левого желудочка повышается индекс Соколова—Лайона (мВ) и суммарный вольтаж зубцов R как в стандартных, так и в левогрудных отведениях. Причем эти признаки более выражены у мужчин.
При анализе ЭКГ необходимо учитывать возрастные особенности детского организма. Так, у детей в возрасте 2—12 лет при оценке гипертрофии желудочков можно пользоваться критериями.
| Гипертрофия левого желудочка | Гипертрофия правого желудочка | ||
| 1. | R aVL > 8 мВ | 1. | R aVR > 4 мВ |
| 2. | R V6 > 25 мВ | 2. | RV4 > 17 мВ |
| 3. | Q V5-6 > 4 мВ при R > 20 мВ | 3. | RV1 + S V5 > 9 мВ |
| 4. | Время внутреннего отклонения (ВВО) в V5 > 0,045 с | 4. | R/SV1 > 4 мВ |
| 5. | Отклонение ЭОС влево | 5. | Форма желудочкового комплекса в виде qR в V1 |
| 6. | Т V5-6 уплощен или отрицателен | 6. | ВВО в V1 > 0,03 с |
| 7. | Отклонение ЭОС вправо > 110о |
При гипертрофии миокарда обоих желудочков диагностика может быть затруднена ввиду того, что признаки гипертрофии того и другого желудочка могут взаимно нивелироваться.
М.К. Осколкова (1986) указывает, что при наличии признаков гипертрофии правого желудочка о гипертрофии обоих желудочков свидетельствуют следующие критерии:
- доминирующий зубец RV5 (высота может быть нормальной) при высоких положительных зубцах Т в этих отведениях;
- увеличение амплитуды зубца q в V5 и V6;
- отклонение ЭОС влево.
При наличии признаков левожелудочковой гипертрофии обнаружение одного из следующих критериев указывает на гипертрофию обоих желудочков:
- доминирующие зубцы R или R’ в отведении V1, причем амплитуда его может быть нормальной;
- амплитуда зубца R больше амплитуды зубца q в отведении aVR;
- зубец S больше зубца R в отведении V6.
На нарушении процессов реполяризации влияет состояние вегетативной нервной системы. Так, нарушения процесса реполяризации миокарда желудочков в покое выявляются у детей с гиперсимпатикотонической вегетативной реактивностью и исходной эйтонией (у 28%).
При функциональных пробах нарушения процесса реполяризации миокарда желудочков определяются у 25% детей с гиперсимпатикотонической и у 50% детей с асимпатикотонической вегетативной реактивностью независимо от исходного вегетативного тонуса.
Выраженность дыхательной аритмии является одним из важных показателей функционального состояния сердца: если колебания длительности интервалов R—R превышают 0,3 с, синусовая аритмия говорит о нарушении регуляции работы синусового узла и может явиться признаком перетренированное™.
Важным есть выявление экстрасистолии. Наряду с экстрасистолией покоя может наблюдаться экстрасистолия непосредственно во время работы и в восстановительном периоде. Принято считать, что экстрасистолическая аритмия, зарегистрированная во время физической нагрузки, указывает на определенное предпатологическое состояние сердечной мышцы. Экстрасистолия покоя считается более благоприятной. Некоторые авторы считают, что исчезновение экстрасистолии, зарегистрированной в покое, при физической нагрузке указывает на “безобидный” характер последней. Однако надо заметить, что опыт спортивной медицины последнего времени, полученный в процессе длительных радиотелеметрических наблюдений, указывает на то, что экстрасистолия является неблагоприятным признаком, независимо от того, зарегистрирована она в покое или при физической нагрузке.
Причины возникновения экстрасистолий у спортсменов весьма разнообразны. Они могут развиваться в результате перенапряжения миокарда, при заболеваниях, вследствие которых наблюдается интоксикация сердечной мышцы, при нарушении нервной регуляции сердечной деятельности, нарушениях минерального обмена и т.д. Необходимо иметь в виду, что у спортсменов при мышечной работе закономерно увеличивается содержание в крови катехоламинов, повышающих возбудимость миокарда.
Субъективно экстрасистолия у спортсменов обычно ощущается в виде “перебоев” в работе сердца. При таких ощущениях необходимо электрокардиографическое исследование. Возникновение экстрасистолий у спортсменов тренер совместно с врачом должны проанализировать в свете индивидуального тренировочного режима.
Предсердно-желудочковая блокада 1 степени (интервал Р—Q больше 0,22 с) наблюдается у спортсменов с выраженным переутомлением или при перетренированности и требует медицинского вмешательства и существенной коррекции тренировочного режима.
Внутрижелудочковая проводимость у спортсменов чаще всего находится на верхней границе нормы, которая соответствует диапазону 0,06—0,09 с. У некоторых спортсменов (особенно с гипертрофией миокарда) длительность комплекса QRS может быть равной 0,1 с.
При выявлении WPW-синдрома необходим тщательный контроль. Часто запрещают занятия ввиду развития опасного нарушение ритма — пароксизмальной тахикардии, недооценивать которое недопустимо.
У спортсменов зачастую присутствует атриовентрикулярная блокада первой степени или второй степени типа Мобитц I, которая отражает главным образом повышенный тонус блуждающего нерва. Также может выявляться увеличение вольтажа зубца Р и комплекса QRS в сочетании с инверсией зубцов Т в боковых отведениях. Удлинение QRS, отклонение электрической оси сердца, наджелудочковые и желудочковые тахикардии не относятся к характерным признакам и могут потребовать проведения дальнейших исследований.
У многих спортсменов наблюдается снижение вольтажа Т-зубцов в стандартных и левых грудных отведениях, а также отмечаются низкие показатели относительного метаболического обеспечения миокарда. Эти изменения особенно выражены при форсированной подготовке в юношеском возрасте.
Детально данные по ЭКГ у спортсменов приведены в книге JI.A. Бутченко и соавт. (1980). По их данным, электрическая ось сердца (ЭОС) у 50% спортсменов нормальная, у 30% — вертикальная, у 4% — отклонение вправо, у 12% — горизонтальная и у 3% — отклонение влево. Вертикальная ЭОС чаще наблюдается у футболистов, лыжников, пловцов, игроков в водное поло и у астеников моложе 20 лет. Горизонтальная ЭОС и отклоненная влево более свойственно борцам, штангистам, бегунам на длинные и средние дистанции и гиперстеникам в 30—49 лет.
Р-Q длиннее у спортсменов, вырабатывающих качество выносливости. Ряд особенностей касается зубцов интервалов и сегментов ЭКГ.
ГИПЕРТРОФИ́Я
ГИПЕРТРОФИ́Я (от гипер… и греч. τροφή – питание), увеличение объёма органа, ткани, клеток, внутриклеточных структур. Встречается как в норме, так и при различных заболеваниях, обеспечивая устойчивость гомеостаза. В основе Г., наряду с увеличением объёма, нередко лежит увеличение и количества структурных единиц (органа, ткани, клетки), т. е. гиперплазия. Напр., в основе Г. паренхиматозного органа лежит Г. паренхиматозных клеток, но при этом может увеличиваться количество элементов стромы, происходить образование новых сосудов (неоангиогенез), обеспечивающее повышенную потребность в питании гипертрофиров. органа. Г. клетки также происходит за счёт увеличения не только объёма её структур, но и их количества. Т. о., Г. и гиперплазия – два неразрывно связанных процесса.
В норме Г. развивается при повышенном функциональном запросе или нейрогормональной стимуляции (напр., Г. миокарда у спортсменов, увеличение объёма матки и молочных желёз во время беременности).
Патологич. Г. может быть истинной (увеличение массы и функциональной активности органа в связи с Г. клеток, обеспечивающих эту активность) и ложной (за счёт разрастания соединит. ткани стромы и жировой ткани при возможной атрофии паренхиматозных клеток).
Истинную Г. подразделяют на рабочую (компенсаторную), викарную (заместительную), регенерационную, нейрогуморальную Г. и гипертрофич. разрастания.
Рабочая Г. развивается при усилении работы органа и характеризуется увеличением объёма его функционально активных структур. Напр., причиной Г. миокарда могут быть разл. гемодинамич. факторы – артериальная и лёгочная гипертензия, врождённые или приобретённые пороки сердца. Масса сердца при этом может превышать нормальные показатели в 3–4 раза. Г. стенки мочевого пузыря развивается у мужчин при гиперплазии предстательной железы, суживающей мочеиспускательный канал и выделяющей гормональные и ростовые факторы, стимулирующие Г. клеток детрузора (мышцы, изгоняющей мочу).
Викарная Г. развивается в одном из парных органов (лёгком, почке, надпочечнике и др.) при гибели или оперативном удалении другого.
Регенерационная Г. формируется в паренхиматозных клетках миокарда, нейронах, гепатоцитах и др. вокруг рубцов при неполной регенерации.
При нейрогуморальной Г. и гипертрофич. разрастаниях, по сути, речь идёт об увеличении объёма органа в осн. за счёт процессов гиперплазии клеточных структур, которые одновременно увеличиваются и в количестве, и в объёме.
Молекулярные механизмы гипертрофии сердца: Часть 1. Физиологическая гипертрофия
Гипертрофия сердца — компенсаторная реакция, возникающая в ответ на увеличение нагрузки на миокард. Физиологическая гипертрофия приводит к усилению функции миокарда: увеличению фракции выброса, усилению сократительной активности.
Выделяют два вида гипертрофии сердца: физиологическую и патологическую, которые отличаются молекулярными механизмами, кардиальным фенотипом и прогнозом для жизни. Последнее — наиболее важно в клинической практике, так как физиологическая гипертрофия помогает «протолкнуть» кровь при более высоких нагрузках, а патологическая ассоциирована с неблагоприятными состояниями, включая инфаркт миокарда, аритмии, и в перспективе может привести к летальному исходу. Также следует заметить, что эти варианты гипертрофии — «конкурентные антагонисты»; между различными сигнальными системами идет постоянная «борьба», результатом которой будет развитие либо физиологической, либо патологической гипертрофии. [1].
При физиологической гипертрофии наблюдается незначительное увеличение массы сердца (на 10–20 %), увеличение кардиомиоцитов (КМЦ) и их пролиферация, при этом фракция выброса сохранена или увеличена. В ткани нет склеротических и некротических изменений, и главное, — физиологическая гипертрофия полностью обратима (за исключением постнатальной) [1].
Механизмы физиологической гипертрофии
Начальные стимулы, приводящие к гипертрофии, не отличаются в случае физиологического или патологического вариантов (например, физическая нагрузка или артериальная гипертензия). Но запускающиеся при этом реакции кардинально различны. Для развития физиологической гипертрофии требуется следующее: увеличение размера клеток, усиление работы митохондрий и продукции энергии, ангиогенез, пропорциональный росту клеток, работа антиоксидантных систем, регуляция пролиферации и регенерации кардиомиоцитов [1].
Физиологическая гипертрофия сердца наблюдается от рождения ребенка и до его взросления (также называется постнатальной гипертрофией), при повышенных физических нагрузках, а также при беременности [2].
Механосенсоры
Механотрансдукция — фундаментальный процесс, сущностью которого является трансформация механических сигналов в биохимические. В сердце существуют сенсорные системы, которые воспринимают механические сигналы (изменения в давлении, объеме и т.д.) и активируют сигнальные системы, ответственные за физиологическую гипертрофию [2].
Ионные каналы с транзиторным рецепторным потенциалом (TRP — transient receptor potential channels) являются одной из таких систем. TRP — это целое суперсемейство трансмембранных белков, которое функционирует как неселективные ионные каналы. Среди них выделяют 7 субсемейств; эти рецепторы отличаются по локализации и своей роли в различных клеточных процессах [3].
Помимо TRP, механочувствительностью обладают различные интегрины. Это трансмембранные белки, способные взаимодействовать с внеклеточным матриксом (и разнообразными его компонентами: фибронектином, ламинином, коллагеном). Реагируя на внеклеточный агент, интегрины могут передавать информацию внутрь клетки путем особых интегрин-ассоциированных белковых комплексов. Это может быть киназа фокальной адгезии (FAK — focal adhesion kinase), интегрин-связанная киназа (ILK — integrin-linked kinase). Последние, в свою очередь, активируют разнообразные сигнальные пути — стандартную протеинкиназу С или PI3K/Act (см. ниже) [2].
Несколько подробнее следует остановиться на интегрин-ассоциированном белке, специфичном для мышечной ткани — мелузине. Он взаимодействует с β1-интегринами, способствуя развитию концентрической гипертрофии с сохранением сократительной активности сердца. Повышение синтеза мелузина инициирует физиологическую гипертрофию и предотвращает ее развитие по патологическому руслу. Верно и обратное: недостаток мелузина приводит к развитию дилатационной кардиомиопатии [1].
И, разумеется, нельзя не сказать о роли Z-линии как механорецептора. Эта часть кардиомиоцита также реагирует на растяжение, передавая сигнал множеству различных белков: телетонину, миопалладину, анкиринам, обскурину… В конечном итоге, все эти процессы оказываются крайне необходимыми для развития физиологической гипертрофии, тогда как дефекты некоторых из этих белков ведут к кардиомиопатиям [2].
Инсулин и инсулиноподобный фактор роста 1
Данные вещества регулируют множество разнообразных клеточных процессов в сердце: пролиферацию, дифференцировку, рост клетки, метаболизм, сократимость и апоптоз. При сердечной недостаточности часто наблюдается резистентность к инсулину. IGF1 (insulin-like growth factor 1) структурно идентичен инсулину; данный агент синтезируется в печени в ответ на СТГ, но также может образовываться и в других органах (в т.ч., в сердце) [1].
Инсулин, как известно, связывается с инсулиновым рецептором — тирозиновой киназой — который фосфорилирует субстраты инсулиновых рецепторов 1 и 2 (IRS1, 2-insulin receptor substrate). IRS в свою очередь активируют сигнальный путь PI3K/Act. Данный сигнальный путь может быть знаком читателю как потенциальная мишень противоопухолевой терапии. PI3K/Act участвует в регуляции клеточного цикла, а в нашем случае обеспечивает рост кардиомиоцитов в ответ на повышение нагрузки на миокард [2], [5].
IGF1 в свою очередь связывается со специфическим рецептором IGF1R (IGF1 receptor) с последующей активацией целого ряда сигнальных путей: RAS–RAF–MEK–MAPK, PLC–IP3R3 и других. Результатом этого также является физиологическая гипертрофия кардиомиоцитов.
Несколько подробнее следует остановиться на PI3K/Act пути (примечательно, что данный сигнальный путь активируется при физической нагрузке). PI3K — фермент-киназа, катализирующий фосфоинозитол-3,4,5-трифосфат. Активация одной из его каталитических субъединиц — p110α — инициирует процессы физиологической гипертрофии и препятствует патологической (как мы помним, данные процессы конкурируют между собой). ACT1 — еще одна киназа, которая активируется фосфоинозитол-3,4,5-фосфатом, и результатом ее работы становится изменение метаболизма клетки. Это достигается путем ингибирования ряда ферментов: гликоген синтазы киназы 3β (данная киназа ингибирует инициирующие трансляцию факторы), fox-O3 (фактор, ингибирующий общий метаболизм белков в клетке) и других. В результате идет активный синтез белка, клетка увеличивается в размере [1].
Трийодтиронин
Т3 — трийодтиронин, гормон щитовидной железы, который влияет как на постнатальную гипертрофию сердца, так и гипертрофию, вызванную физическими нагрузками (хотя последнее — не так достоверно) [2].
Сразу после рождения концентрация Т3 в крови существенно увеличивается. Гормон связывается с рецепторами TRα и TRβ (thyroid hormone receptors), что приводит к «переключению» транскрипции гена MYH7 на MYH6 (данные гены кодируют последовательности тяжелых цепей миозина — myosin heavy chains). MYH7 кодирует β-тяжелые цепи миозина, MYH6 — α-изоформу [1]. Это крайне важно, так как α-изоформа обладает большей АТФазной активностью и, как следствие, клетка с активным MYH6 способна сокращаться сильнее [2], [6].
Взаимодействие Т3 с рецепторами к ретиноевой кислоте (ядерные рецепторы стероидных и тиреоидных гормонов, прим.) приводит к увеличению экспрессии кальциевой АТФазы-2 (SERCA2 — sarcoplasmatic/endoplasmatic reticular calcium ATPase-2), ингибируя MYH7 и экспрессию фосфоламбана (белок в кардиомиоцитах, который супрессирует SERCA2) [2].
SERCA2 — эволюционно наиболее древняя изоформа кальциевых АТФаз млекопитающих. Всего таких изоформ три (с подтипами). SERCA2 имеет три подтипа: a, b и с; для сердца наиболее специфична SERCA2а (97,5 %), также имеется небольшое количество SERCA2b (2,5 %). Функция данного белка состоит в контроле цитозольного Ca2+ и, как следствие, в регуляции сократительной функции всего кардиомиоцита [7].
Фосфоламбан, работу которого ингибирует Т3, является белком-регулятором SERCA2 (точнее, наиболее изученным из этих регуляторов). В своем активном — дефосфорилированном — состоянии фоcфоламбан супрессирует SERCA2 и приводит к снижению концентрации Ca2+ в цитоплазме, что, в свою очередь, снижает сократительную способность кардиомиоцита [2], [7].
Помимо этого, Т3 также активирует транскрипцию β1-адренорецепторов, белков натриевых и кальциевых каналов, сердечного тропонина I, белков натрий/кальциевой помпы и аденилатциклазы V и VI типов [1], [2]. Также гормон увеличивает содержание TRα1, который активирует уже известный нам PI3K [2].
Конечным результатом работы трийодтиронина в кардиомиоцитах является усиление сократительной активности миокарда и кардиопротекция [1].
Оксид азота (NO)
Физическая нагрузка стимулирует β3-адренорецепторы в эндотелиоцитах, в ответ активируется эндотелиальная NO-синтаза (NOS3), которая синтезирует собственно оксид азота — известное вазоактивное вещество. После этого происходит следующее:
NО активирует растворимую гуанилатциклазу → повышается содержание цГМФ → активируется цГМФ-зависимая протеинкиназа G (PKG) → PKG фосфорилирует два белка-регулятора (RGS2 и RGS4), которые ингибируют G-связанные рецепторы, ответственные за развитие патологической гипертрофии.
Такая схема нужна для того, чтобы гипертрофия развивалась по физиологическому варианту. Дефект либо β3-адренорецепторов, либо NOS3, либо белков RGS2 и RGS4 ведет к разрыву всей цепи кардиопротекции, что вполне может закончиться инфарктом миокарда [1].
Ангиогенез
Для нормального функционирования клеткам нужно достаточное кровоснабжение. Если плотность сосудов увеличивается соразмерно росту кардиомиоцитов — развивается физиологическая гипертрофия. Но когда клетки растут быстрее, чем того может позволить капиллярная сеть — возникает хроническая гипоксия.
Одним из важнейших факторов, определяющих ангиогенез, является сосудистый эндотелиальный фактор роста (VEGF — vascular endothelial growth factor). VEGF также может быть знаком читателю — ингибиторы данного фактора используются при лечении различных видов рака. Однако подобные препараты обладают существенной кардиотоксичностью и могут привести к различным кардиомиопатиям (что косвенно свидетельствует о важности VEGF для функционирования миокарда). Помимо прочего, изменение уровня VEGF также связано с перипартальной (послеродовой) кардиомиопатией [1], [2].
В начале статьи упоминались сигнальные молекулы NFAT (в разделе «механосенсоры»). VEGF, влияя на эндотелиоциты, действуют именно через систему NFAT, вызывая усиленный ангиогенез. Таким образом, данные нуклеарные факторы, помимо своей антиапоптотической роли, влияют и на кровоснабжение кардиомиоцитов [8].
Помимо VEGF, важную роль оказывает и тромбоцитарный фактор роста (PDGD — platelet-derived growth factor). Доподлинно неизвестны механизмы, которыми осуществляется влияние PDGD на ангиогенез; однако дефект данного фактора в экспериментах на мышах приводит к сердечной недостаточности и ухудшению кровоснабжения [1].
Также нельзя не упомянуть о еще одном важнейшем веществе. Гипоксия-индуцируемый фактор 1α (HIF1α — hypoxia-inducible factor) — это главный фактор транскрипции, обеспечивающий постоянное снабжение клеток кислородом путем регуляции ангиогенеза, модификации сосудов и регуляции метаболизма глюкозы. В случае патологической гипертрофии антиген р53 (известный своей связью с развитием опухолей) активирует убиквитинирование и протеасомную деградацию HIF1α [1]. В норме же HIF1α активирует различные сигнальные молекул (в т.ч. VEGF) и способствует усиленному ангиогенезу.
Нейрегулин-1
Нейрегулины (1-4) — это члены семейства эпидермальных факторов роста. В сердечно-сосудистой системе представлен в основном нейрегулин-1. Сигнальный путь этого фактора (и его рецепторов) играет важную роль в адаптации миокарда к нагрузке и формировании сердечной мышцы.
Нейрегулин-1 действует на киназу семейства ErbB (группа молекул, структурно схожих с эпидермальным фактором роста; белки данного семейства часто рассматриваются как потенциальные онкомаркеры, прим.). Нейрегулин-1 активирует ErbB2 и ErbB4, что приводит к стимуляции уже упоминавшейся PI3K-системы. Результатом этого взаимодействия является пролиферация кардиомиоцитов, что предотвращает ишемические поражения [1].
МикроРНК и РНК-связывающие белки
МикроРНК (мкРНК) экспрессируются в сердце при физиологической гипертрофии, индуцированной аэробной физической нагрузкой. Например, мкРНК-222 у мышей ингибирует четыре мишени, потенциально ответственные за декомпенсацию и развитие сердечной недостаточности: р-27 (кодирует ингибитор клеточного цикла), Hipk1 и Hipk2 (кодируют протеиновые киназы), Hmbox1 (кодирует ингибитор транскрипции). Иными словами, активация мкРНК-222 способствует росту и пролиферации кардиомиоцитов [1].
Также любая форма гипертрофии нуждается в синтезе белка de novo. И здесь задействованы эукариотический фактор трансляции 4F (elF4F) и мишень рапамицина млекопитающих mTORC1 — вместе они запускают усиленный синтез белка. Помимо этого, в ответ на физиологический (и патологический) стимул в клетках миокарда происходит элонгация поли-А последовательности и экспрессия специфического полиаденилат-связывающего белка 1 (PABPC1 — polyadenylate-binding protein 1), который связывается с elF4F и таким образом способствует накоплению в клетках различных строительных (и не только) белков. Повышенная экспрессия PABPC1 в кардиомиоцитах необходима для физиологической гипертрофии (о роли белка в патологическом ее варианте пока что ничего не известно) [1].
Источники:
- M. Nakamura and J. Sadoshima, ‘Mechanisms of physiological and pathological cardiac hypertrophy’, Nat. Rev. Cardiol., vol. 15, no. 7, pp. 387–407, 2021.
- M. Maillet, J. H. Van Berlo, and J. D. Molkentin, ‘Molecular basis of physiological heart growth: Fundamental concepts and new players’, Nat. Rev. Mol. Cell Biol., vol. 14, no. 1, pp. 38–48, 2013.
- B. Nilius and G. Owsianik, ‘The transient receptor potential family of ion channels’, Genome Biol., vol. 12, no. 3, 2011.
- W. T. Pu, Q. Ma, and S. Izumo, ‘NFAT transcription factors are critical survival factors that inhibit cardiomyocyte apoptosis during phenylephrine stimulation in vitro’, Circ. Res., vol. 92, no. 7, pp. 725–731, 2003.
- M. Osaki, M. Oshimura, and H. Ito, ‘PI3K-Akt pathway: Its functions and alterations in human cancer’, Apoptosis, vol. 9, no. 6, pp. 667–676, 2004.
- E. M. McNally, R. Kraft, M. Bravo-Zehnder, D. A. Taylor, and L. A. Leinwand, ‘Full-length rat alpha and beta cardiac myosin heavy chain sequences’, J. Mol. Biol., vol. 210, no. 3, pp. 665–671, 1989.
- P. Vangheluwe, L. Raeymaekers, L. Dode, and F. Wuytack, ‘Modulating sarco(endo)plasmic reticulum Ca2+ ATPase 2 (SERCA2) activity: Cell biological implications’, Cell Calcium, vol. 38, no. 3-4 SPEC. ISS., pp. 291–302, 2005.
- T. A. Zaichuk, E. H. Shroff, R. Emmanuel, S. Filleur, T. Nelius, and O. V. Volpert, ‘Nuclear factor of activated T cells balances angiogenesis activation and inhibition’, J. Exp. Med., vol. 199, no. 11, pp. 1513–1522, 2004.